Uso de bioinformática en el
diagnóstico de hemoglobinopatías
Rosenfeld-Mann, Dra. Rocio Trueba-Gómez.
Coordinación de Hematología Perinatal. Instituto Nacional de Perinatología “Isidro Espinosa de los
Reyes”. CDMX. México
Introducción
En adultos, la hemoglobina A está formada por 4 subunidades, dos alfas y dos betas. El gen de las subunidades β (HBB), está localizado en 11p15.4 (GRCh38.p13, NC_000011.10 (5225464 - 5227071, complemento)) tiene 3 exones, transcribe seis productos y cuatro isoformas, dos de 147 y otras 2 de 99 y 111 aminoácidos. Hay reportadas 152 variantes genéticas con importancia clínica (OMIM #141900).
Las hemoglobinopatías son defectos en la estructura y/o síntesis de las cadenas globínicas causadas por mutaciones en sus genes, entre ellas las talasemias β y/o α. Las talasemias son enfermedades monogénicas1, que pueden presentarse inclusive cuando se afecta un solo alelo.
Las sustituciones en los residuos aminoacídicos resultado de mutaciones genéticas pueden alterar la estructura terciaria de la proteína final y por tanto su funcionalidad, sin embargo, dicha proteína puede no variar significativamente en su relación tamaño/peso y pH. Algunas de las hemoglobinas anormales no pueden ser detectadas por medio de la electroforesis, y muchas de ellas solo se identifican por isoelectroenfoque (IEF) posterior a la esplenectomía. Estas observaciones sugieren que las variantes de la cadena β son altamente inestables y se degradan rápidamente después de la síntesis2, 3.
Una vez obtenida la secuencia, es
posible modelar la estructura 3D, y
predecir las implicaciones que las
sustituciones pudiesen tener en la
funcionalidad final
posible modelar la estructura 3D, y
predecir las implicaciones que las
sustituciones pudiesen tener en la
funcionalidad final
Otra herramienta frecuentemente utilizada para la identificación de las variaciones genéticas es PCR - tiempo real, no obstante, la alta afinidad de los iniciadores solo puede identificar las variantes sobre las que fueron diseñados, ignorando alguna otra cercana, dando resultados falsos negativos.
Particularmente, para el análisis de variantes de la cadena β, hemos propuesto una estrategia de secuenciación Sanger basada en dos pares de iniciadores para amplificar y secuenciar completo el gen HBB4.
Una vez obtenida la secuencia, es posible modelar la estructura 3D, y predecir las implicaciones que las sustituciones pudiesen tener en la funcionalidad final. Muchas herramientas bioinformáticas están disponibles en línea y son gratuitas.
Material y métodos
El caso que se presenta, es el producto de una madre que ingresó al instituto por edad de riesgo y preeclamsia. Sin antecedentes de hemoglobinopatías. Desconocemos los antecedentes paternos. El neonato fue a término con niveles normales de ferritina sérica, Hb e índices eritrocitarios. A los dos días de vida extrauterina se diagnosticó con hiperbilirrubinemia multifactorial y fue tratado con fototerapia.
En ese momento la madre refiere que el padre siempre se veía pálido y cansado. Se solicita interconsulta al servicio de hematología perinatal.
Electroforesis capilar
Se utilizó el Capillarys 2 flex piercing, con el kit “The capillarys hemoglobin(E)” y control HBAF, (Sebia Lisses, Francia).
Extracción de DNA
Se extrajo el DNA a partir de muestras de sangre con EDTA, utilizando el reactivo High Pure PCR Template Preparation Kit (Roche Diagnostic, Germany) de acuerdo a las condiciones del fabricante. Se almacenaron a -70˚C hasta su análisis.
PCR - tiempo real
Como parte de los estudios moleculares de rutina para identificar variantes de hemoglobina y síndromes talasémicos, realizamos un tamiz del gen HBB (HGNC:4827) por PCR - tiempo real de 9 SNP (rs334, rs33931746, rs34563000, rs63749819, rs34856846, rs281864897, rs63750504, rs33946267, rs33971440) mediante sondas de hibridación (LightCycler FastStar DNA Master HybProbe. Roche Cat. 03003248001), en un termociclador LightCycler 2.0 (Roche Diagnostic, Germany).
Secuenciación Sanger
Diseñamos dos pares de iniciadores: uno que incluye la región promotora, exón 1 y exón 2 y otro par para el exón 3 de HBB4. Para la PCR utilizamos HotStarTaq DNA Polymerase (Qiagen, Hilden, Germany Cat. No 203445) por 35 ciclos.
Para la secuenciación utilizamos BigDye™ Terminator v3.1 Cycle Sequencing Kit (Thermofisher Cat. No. 4337455). En un 3500 Genetic Analyzer de Applied BioSytem.
Secuencias de referencia
Utilizamos como referencia del gen de HBB las entradas ENSG00000244734.4 y 3043 (Ensembl y NCBI/gene respectivamente). Las de los transcritos fueron ENST00000335295.4 y NM_000518.5 (Ensembl y NCBI/nuccore respectivamente). Las secuencias de la proteína fueron NP_000509.1 (NCBI/protein) y P68871 (Uniprot).
Nomenclatura
Para la nomenclatura de las variantes seguimos las recomendaciones de la Human Genome Variation Society (HGVS) (http://www.hgvs.org/mutnomen) y corroboramos la sintaxis correcta utilizando Mutalyzer (https://mutalyzer.nl).
Análisis Bioinformático
Para el diseño de primer, análisis, alineamiento, comparación de las secuencias, extracción de las secuencias codificantes y traducción utilizamos el software Geneious® 7.1.9 (https://www.geneious.com)
Predicción
La predicción in silico de las consecuencias de las variaciones se realizó con las herramientas on line PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)5, SIFT Sequence (https://sift.bii.a-star.edu.sg/www/SIFT_seq_submit2.html)6, 7, Provean protein (http://provean.jcvi.org/seq_submit.php) y MutationTaster (http://www.mutationtaster.org/)8.
Estructura 3D
Se identificaron los marcos de lectura con Expasy (https://www.expasy.org/resources/translate). Los modelos 3D se elaboraron con Raptorx (http://raptorx.uchicago.edu/)9.
Este estudio fue aprobado por los comités de investigación, ética y bioseguridad del Instituto Nacional de Perinatología (212250-3101-10808-02-15).
Resultados
La electroforesis realizada en la muestra del neonato fue normal para su edad, muestra predominio de la hemoglobina fetal y sin alteraciones aparentes en la hemoglobina A. (Figura 1)
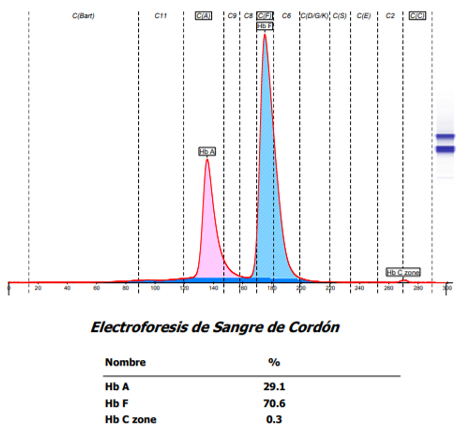
Figura 1. Electroferograma de la muestra de hemoglobina del neonato
Se procedió a realizar el tamiz del gen HBB por PCR - tiempo real para las variantes frecuentemente analizadas en población mexicana. El binomio madre-hijo fue wild type para todo el panel.
Secuenciamos el gen HBB completo en busca de otras variantes. La secuencia de la madre no tuvo alteraciones. Sin embargo, en el neonato, mediante la secuenciación y análisis bioinformático, se encontró una doble mutación en el mismo alelo (HBB: c.266T> G) y (HBB: c.282T> C). (figura 2)
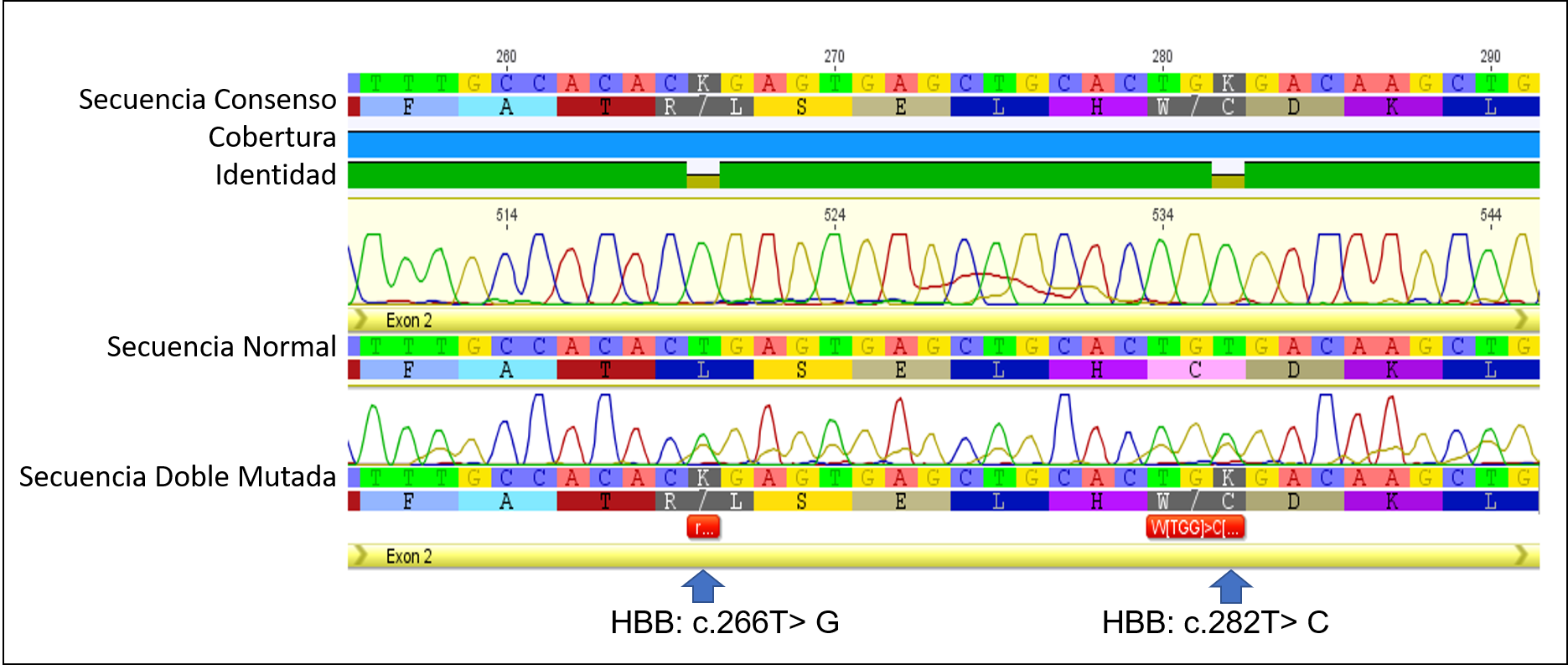
Figura 2. Electroferogramas de las secuencias consenso, normal y doble mutada (DNA/Proteína). Las flechas muestran la localización de las variaciones.
Estos cambios dan lugar a las variantes Hb Borås y Hb Santa Giusta Sardegna previamente reportadas por otros grupos; sin embargo, no hay reportes de su aparición combinada en el mismo alelo. Las variantes p.Leu88Arg y p.Cys93Trp no alteran la longitud final de la proteína, los resultados bioinformáticos sugieren que existen diferencias en la estructura terciaria de la β-globina, afectando principalmente a las hélices E y F, que son los sitios de interacción con el grupo hemo. Por medio de estudios in silico, predijimos la estructura terciaria de la sub unidad β-globina para la forma WT (Figura 3a) y la secuencia doble mutada (Figura 3b).
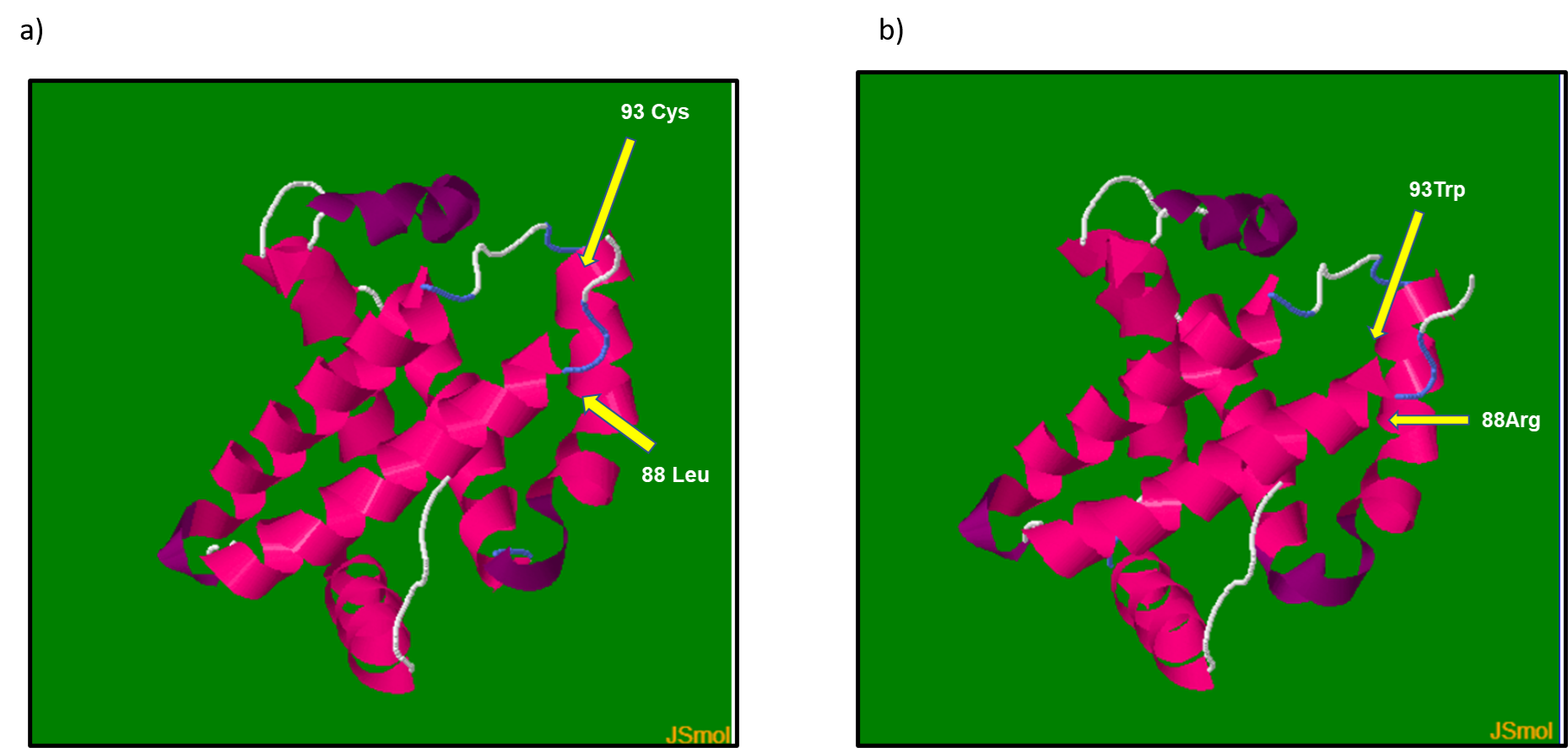
Figura 3. Modelos 3D de a) Hemoglobina Normal y b) Hemoglobina con doble mutación.
Determinamos el número de residuos y la longitud en nm de las hélices E y F. Calculamos las amplitudes de cinco vectores. Las primeras tres considerando como origen la histidina 63 distal (H63) hacia el primer aminoácido de la hélice F, a la histidina 92 proximal y al último aminoácido de la hélice F. La cuarta distancia fue del primer aminoácido de la hélice F al primero de la E y la quinta entre los últimos aminoácidos de las hélices mencionadas. Los valores fisicoquímicos teóricos obtenidos muestran que las dos proteínas tienen 147 residuos con escisión de la metionina inicial, La proteína normal tiene un peso molecular de 15.998 kDa, punto isoeléctrico de 7.32 y un coeficiente de extinción de 15,595, en cambio la proteína mutada tiene un peso molecular de 16.125 kDa, con un punto isoeléctrico de 7.87 y un coeficiente de extinción 20,970. Los algoritmos predictivos in silico
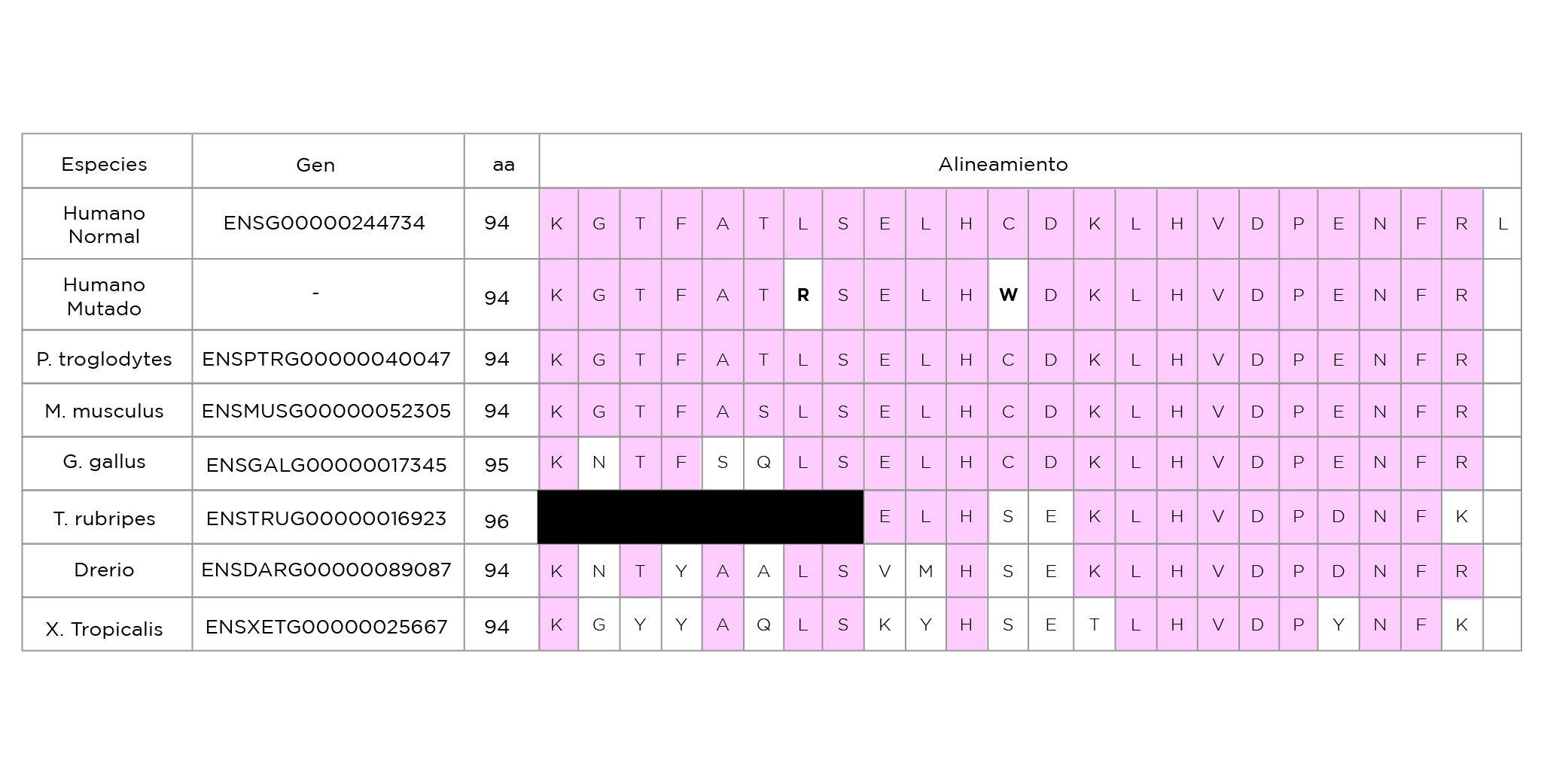
clasifican a la proteína doble mutada como patogénica debido a posibles alteraciones funcionales y estructurales. La comparación filogenética de las secuencias de la β-globina de diferentes especies muestran una alta conservación en los residuos aminoacídicos en los puntos de sustitución. (Tabla 1)
Tabla 1. Comparación de la secuencia de la β-globina de varias especies, en la región de la Cys94.
Discusión
Las variantes Hb Borås10 y Hb Santa Giusta Sardegna11 están reportadas al menos en HBVar (419 y 2837), sin embargo, no hay reportes de su aparición combinada en el mismo alelo.
Un error en el que podemos caer, es interpretar el conjunto de mutaciones encontradas como una suma aritmética de las consecuencias de una mutación más otra; se trata en realidad de una consecuencia diferente; en este caso son dos cambios en una región importante de la sub unidad β, que por ser una combinación nueva y por la baja frecuencia de la segunda variante, no se identificó por nuestro panel de PCR - tiempo real.
La variante compuesta no afecta la longitud de la proteína. La primer sustitución es una Leu en el residuo 88 por una Arg, es una variante patogénica misense descrita por primera vez en 196910. Siempre se había reportado como una mutación simple. La otra mutación es una sustitución de un Trp en 93 en lugar de una Cis11. Por si sola, esta nueva variante se predice patogénica. Stamler y colaboradores demostraron que in vitro e in vivo la HbA se somete a S-nitrosilación (SNO-HbA), que el sitio de S-nitrosilación es probablemente el grupo tiol reactivo de Cys93 de β-globina12-14. Las dos mutaciones traducen a aminoácidos dentro de la hélice F. En la proteína normal la hélice F está formada por 13 residuos y tiene una longitud de 1.53nm, mientras que la proteína mutada tiene 14 residuos, su longitud es de 1.951nm, es decir 27.516% más larga. En la primera sustitución originalmente es una Leucina, un aminoácido con cadena lateral no polar (un grupo isobutilo) y masa molar 131.17 g/mol y se cambia por una arginina que tiene un grupo guanidino, y por lo tanto cuando se ioniza tiene menor densidad de carga que otros aminoácidos como la lisina, y mayor que la histidina. La segunda sustitución originalmente es una cisteina (121.16 g/mol), que contiene un grupo tiol (-SH) en su cadena lateral, por lo que considera polar e hidrófilo, el tiol es susceptible a la oxidación y da lugar a puentes disulfuros que son importantes para la estructura de muchas proteínas. Se sustituye por triptofano de masa molecular 204.23 g/mol, se clasifica entre los aminoácidos apolares (hidrófobos). Se caracteriza por una cadena lateral con el grupo indol. Comparando la secuencia de la β-globina de varias especies, en estas posiciones en particular, podemos ver que es una secuencia altamente conservada y que las variaciones afectarán la estabilidad del tetrámero.
Con todo esto, es posible proponer que se trate de una nueva forma de hemoglobina. No fue posible describir clínicamente las consecuencias de estos cambios, ya que, el neonato fue dado de alta sin seguimiento pediátrico y no se había concluido el análisis molecular.
Conclusión
Los cambios en la secuencia del gen no siempre tienen consecuencia sobre la estructura de la proteína. Sin embargo, en nuestro estudio, encontramos dos mutaciones que han sido descritas como patogénicas de manera individual. El haplotipo del neonato, las referencias dadas por la madre de la posible anemia paterna, y las predicciones in silico, indican que el niño podría cursar con talasemia. Desgraciadamente no fue posible contactar a la madre para dar el seguimiento. Esta estrategia nos permitirá plantear estrategias y establecer un diagnóstico temprano de talasemias.
BIBLIOGRAFÍA
1. Mettananda S, Higgs DR. Molecular Basis and Genetic Modifiers of Thalassemia. Hematol Oncol Clin North Am. 2018;32(2):177-91.
2. Thom CS, Dickson CF, Gell DA, Weiss MJ. Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harb Perspect Med. 2013;3(3):a011858.
3. Thein SL. Pathophysiology of beta thalassemia--a guide to molecular therapies. Hematology Am Soc Hematol Educ Program. 2005:31-7.
4. Martinez Villegas O, Mendoza-Melendez D, Trueba-Gomez R, Rosenfeld-Mann F, Baptista-Gonzalez HA, Estrada-Juarez H. Analysis of a Novel Mexican Variant of the HBB Gene Associated with beta-Thalassemia Using Bioinformatic Tools. Hemoglobin. 2021;45(2):87-93.
5. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248-9.
6. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic acids research. 2012;40(Web Server issue):W452-7
7. Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11(1):1-9.
8. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361-2.
9. Ma J, Wang S, Zhao F, Xu J. Protein threading using context-specific alignment potential. Bioinformatics. 2013;29(13):i257-65.
10. Hollender A, Lorkin PA, Lehmann H, Svensson B. New unstable haemoglobin boras: beta 88 (F4) leucine-arginine. Nature. 1969;222(5197):953-5.
11. Fais A, Sollaino MC, Barella S, Perseu L, Era B, Corda M. A new beta chain hemoglobin variant with increased oxygen affinity: Hb Santa Giusta Sardegna [beta93(F9)Cys-->Trp; HBB c.282T>G]. Hemoglobin. 2012;36(2):151-6.
12. Chan NL, Rogers PH, Arnone A. Crystal structure of the S-nitroso form of liganded human hemoglobin. Biochemistry. 1998;37(47):16459-64.
13. Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380(6571):221-6.
14. Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, et al. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997;276(5321):2034-7