Genética en miocardiopatía dilatada
Las miocardiopatías son un grupo heterogéneo de enfermedades del miocardio, asociadas a disfunción mecánica y/o eléctrica, que generalmente presentan una inadecuada hipertrofia, dilatación o restricción ventricular y pueden deberse frecuentemente, a causas de origen genético. La Sociedad Europea de Cardiología (ESC) ha clasificado las causas de Miocardiopatía Dilatada (MCD) como familiares (genéticas) o no familiares (no genéticas). Por otro lado, la American Heart Association (AHA), clasifica las causas como: genéticas, adquiridas (exposición a toxinas, infecciones, enfermedades metabólicas, etc.) ó mixtas (la predisposición genética interactúa con factores extrínsecos o ambientales desencadenando el fenotipo). En la mayoría de los estudios epidemiológicos, la proporción de pacientes con MCD determinada genéticamente se subestima sustancialmente debido a la presentación clínica variable, la penetrancia incompleta de la enfermedad y la falta de fenotipos específicos. La etiología de origen desconocido o ¨idiopática¨ se presenta hasta en un 30% de los casos, a su vez, la MCD idiopática tiene una etiología genética con presentación familiar hasta en un 50% de los casos e involucran genes que codifican un grupo heterogéneo de moléculas que participan en la generación y transmisión de la fuerza contractil, integridad del sarcómero, arquitectura citoesquelética y nuclear, homeostasis electrolítica, función mitocondrial y transcripción. Los enormes avances en los campos del diagnóstico genético y la biología molecular, han permitido que cada día sea posible identificar en más pacientes las causas específicas de la MCD, por lo que la denominación ¨idiopática¨ debería tener, progresivamente, un uso más restringido. El diagnóstico genético también puede ayudar a predecir el pronóstico, especialmente con respecto al riesgo de arritmias ventriculares y muerte súbita para ciertas mutaciones. Además, el estudio en familiares de la variante identificada en el caso índice, permite identificar a quienes están en riesgo o con la enfermedad en una etapa temprana, ofreciendo la oportunidad de una intervención precoz. En los últimos años, se han identificado más de 120 genes implicados y/o potencialmente asociados al desarrollo de MCD. Un estudio genético completo permitiría identificar la causa de la enfermedad en aproximadamente un 50% de los casos que no tienen otra etiología identificable (MCD idiopática). Esta rentabilidad diagnóstica depende de cada caso en particular y se ha descrito en algunas series, que puede superar el 70% cuando la enfermedad tiene una presentación familiar.
Los resultados del estudio genético pueden ser de una valiosa ayuda para el médico tratante, aportando información a nivel diagnóstico, pronóstico (especialmente en subgrupos de pacientes con alto riesgo de arritmia ventricular y muerte súbita) y terapéutico (medidas preventivas, indicación precoz de dispositivos, etc.). Adicionalmente, las pruebas genéticas en los familiares de los individuos afectados, permiten identificar a portadores que están en riesgo o con expresión de la enfermedad en etapa temprana, ofreciendo la oportunidad de una intervención anticipada.
El resultado del estudio genético puede aportar información sobre el momento más adecuado para el implante de un desfibrilador como prevención primaria en MCD. Es sabido que, en la MCD como grupo general, el punto de corte para decidir el implante de desfibrilador como prevención primaria es una fracción de eyección del ventrículo izquierdo igual o menor que el 35%. Sin embargo, múltiples estudios han demostrado que existen distintas mutaciones asociadas con un riesgo incrementado de eventos arrítmicos y muerte súbita, que pueden aparecer con deterioros mucho menores de la fracción de eyección y/o con leves dilataciones ventriculares.
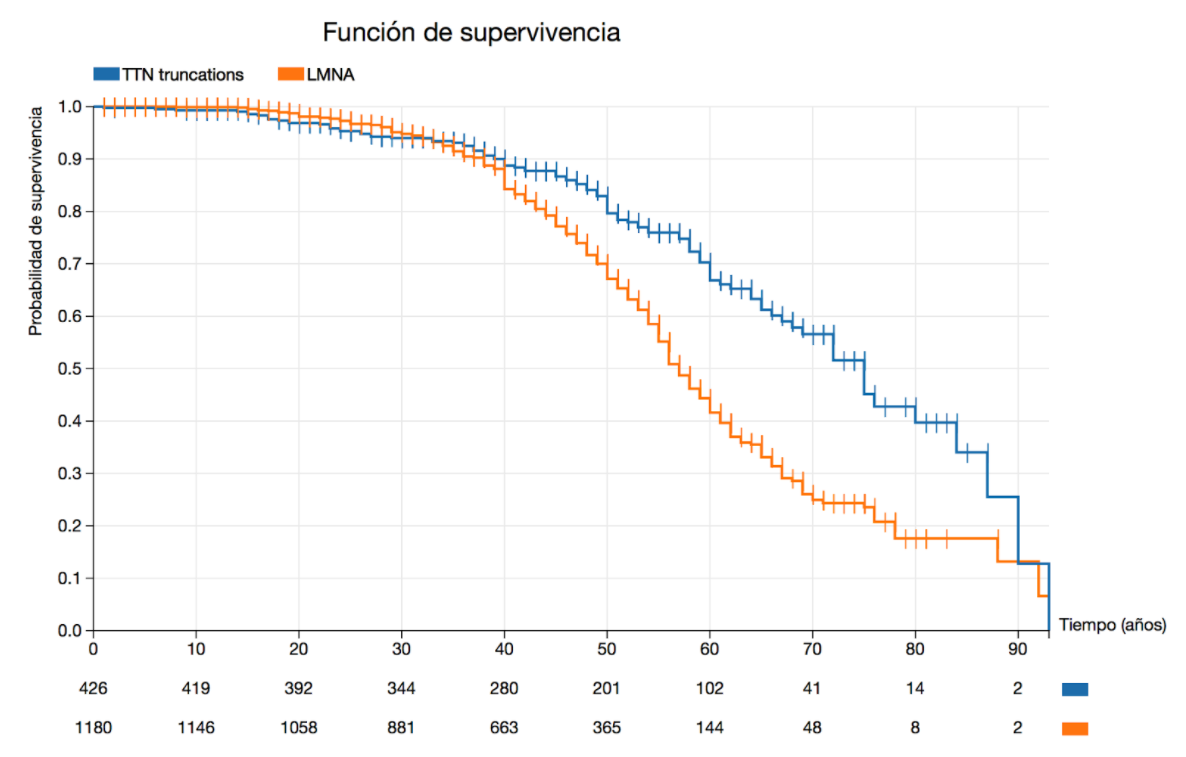
En las guías de práctica clínica vigentes hay ejemplos sobre cómo el conocimiento de la variante genética causante de la enfermedad puede modificar radicalmente la aproximación clínica a los pacientes afectados. Las variantes patogénicas en Lámina A / C (codificada por el gen LMNA) representan el ejemplo de cómo la identificación de una mutación específica puede cambiar el tratamiento de rutina, al obtener una indicación de clase IIa para el implante de DAI en presencia de ciertos factores de riesgo adicionales, independientemente de la gravedad de la disfunción ventricular izquierda. En el gráfico 1, se puede observar una curva realizada por el método de Kaplan-Meier que compara la supervivencia en portadores de variantes patogénicas en LMNA respecto a portadores de variantes patogénicas en TTN. Se observa que la incidencia de eventos es significativamente mayor a partir de la 4ta década de la vida (p=0.000108) en portadores de variantes patogénicas en LMNA.
Los eventos cardiovasculares en los portadores de variantes en LMNA son principalmente a expensas de muerte súbita, en cambio, los portadores de variantes patogénicas en TTN, suelen sufrir eventos de muerte por fallo cardíaco.
Referencias
1. Rev Esp Cardiol 2000; 53: 360-393
Cardiovascular Quality and Outcomes 2021; 14 :476-569.
2. McKenna WJ, Judge DP. Epidemiology of the inherited cardiomyopathies. Nat Rev Cardiol . 2021;18:22–36.
3. Barriales-Villa R, Gimeno-Blanes JR, Zorio-Grima E, Ripoll-Vera T, Evangelista-Masip A, Moya-Mitjans A, et al. Plan of Action for Inherited Cardiovascular Diseases: Synthesis of Recommendations and Action Algorithms. Rev Española Cardiol. 2016;69:300–9.
4. Fatkin D, Huttner IG, Kovacic JC, Seidman JG, Seidman CE. Precision Medicine in the Management of Dilated Cardiomyopathy. J Am Coll Cardiol . 2019;74:2921–38.
5. Escobar-Lopez L, Ochoa JP, Mirelis JG, Espinosa MÁ, Navarro M, Gallego-Delgado M, et al. Association of Genetic Variants With Outcomes in Patients With Nonischemic Dilated Cardiomyopathy. J Am Coll Cardiol . 2021;78:1682–99.
6. van Rijsingen IAW, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, et al. Risk Factors for Malignant Ventricular Arrhythmias in Lamin A/C Mutation Carriers. J Am Coll Cardiol . 2012;59:493–500.
7. Herman DS, Lam L, Taylor MRG, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of Titin Causing Dilated Cardiomyopathy. N Engl J Med . 2012;366:619–28.