El uso apropiado de herramientas hematológicas para el diagnóstico de una familia con S-Beta Talasemia
Laboratorio de Biología Molecular y Celular, Instituto de Hematopatología, México.
Introducción
Las talasemias son las enfermedades hereditarias más comunes del ser humano, y se caracterizan por defectos cuantitativos en la síntesis de hemoglobina.
Aproximadamente una de cada mil o dos mil personas en el mundo tienen talasemia, la mayor parte de ellos con formas menos graves (menores), mientras que las formas más graves (intermedias o mayores) generalmente se presentan en sujetos homocigotos o dobles heterocigotos y son, por lo tanto, más raras.
El término talasemia es un neologismo médico acuñado en inglés (thalassemia), derivado del griego antiguo thalassa = mar y haima = sangre, lo que quiere decir literalmente “sangre de los que viven próximos al mar”. Para los antiguos pobladores, era el Mar Mediterráneo; y éstas enfermedades son comunes (aunque de ninguna manera exclusivas) en países que bordean dicho mar.
La hemoglobina (Hb) es una proteína tetramérica compuesta por diferentes combinaciones de subunidades de globinas; cada subunidad de globina está asociada con un grupo hemo, que puede transportar una molécula de oxígeno.
El problema primario en este grupo de enfermedades es la disminución o ausencia de síntesis de cadenas de globinas por mutaciones en los genes que codifican para dichas proteínas.
Las talasemias se clasifican dentro del grupo de anemias microcíticas hipocrómicas, aunque la disminución del volumen globular medio y de la concentración media de hemoglobina globular dependen del nivel de reducción en la síntesis de las cadenas de globinas. Además de ser microcíticas hipocrómicas existe un componente hemolítico en las talasemias que se manifiesta por el aumento variable de reticulocitos.
En las talasemias “menores” (generalmente heterocigotos), menos graves, los pacientes tienen microcitosis notable pero con poca hipocromía, y los niveles de hemoglobina sólo están ligeramente disminuidos o incluso pueden ser normales.
Por otra parte, en las talasemias intermedias y mayores (generalmente homocigotos o dobles heterocigotos), los niveles de hemoglobina son más bajos y la microcitosis e hipocromía son notables; en éstas, la cantidad de reticulocitos es mayor que en las formas menores.
Uno de los objetivos fundamentales al clasificar un caso de talasemia, es definir cuál es el tipo de cadena globínica disminuida, por ejemplo beta-talasemia, significa que hay disminución o ausencia de cadenas beta mientras que en las alfa-talasemias existe disminución o ausencia de cadenas alfa, etc.
Como los genes que codifican para las globinas están localizados en cromosomas autosómicos; hombres y mujeres tenemos cuando menos un par de genes para cada cadena, ya que para las cadenas alfa existen 4 genes (dos en cada cromosoma 16) y para las cadenas gamma existen también 4 genes (dos en cada cromosoma 11). Para el resto de las globinas (beta, delta, épsilon) existen dos genes para cada una localizados en los cromosomas 11 y para las cadenas zeta existen dos genes localizados en los cromosomas 16.
El factor más importante que determina la gravedad de una talasemia es el número de genes afectados. Un gen talasémico es aquel que tiene mutaciones de diversos tipos que al final llevan a la producción de una cantidad menor de globina o ausencia de ésta. Por ejemplo, un paciente que tiene un gen beta talasémico, pero que tiene el otro gen beta normal (le llamamos heterocigoto), tiene una forma leve de beta-talasemia. Aunque sus eritrocitos son pequeños (no crecen porque no tienen suficiente hemoglobina) e hipocrómicos (tienen incluso menos hemoglobina de la esperada para su pequeño volumen), la médula ósea produce muchos eritrocitos y los niveles de hemoglobina no se encuentran muy disminuidos. Decimos que estos pacientes tienen talasemia menor: más de 10 g de hemoglobina / dL, o incluso a veces niveles normales.
En cambio los pacientes homocigotos o dobles heterocigotos pueden tener talasemia mayor o intermedia, dependiendo de qué tanta disminución en la síntesis de beta globina se produzca según el tipo de mutación. Por ejemplo, los pacientes homocigotos para talasemia beta “cero” (genes que no producen nada de cadenas globínicas beta) tienen talasemia mayor: menos de 5 g de hemoglobina / dL, muchos poiquilocitos y eritroblastos en los frotis, así como dependencia absoluta de transfusión.
En los casos de alfa talasemia el problema es más complejo porque hay 4 genes para alfa globinas. Los pacientes que poseen sólo un gen alfa talasémico tienen talasemia “mínima”, los que poseen dos, tienen talasemia menor, los que poseen tres. tienen talasemia intermedia y los que poseen cuatro genes alfa talasémicos no llegan a término o mueren al nacimiento con una anemia muy grave.
Volviendo a las beta talasemias, en estado heterocigoto la talasemia es menor, en muchos de estos casos la enfermedad es asintomática. La biometría hemática presenta mínima o nula anemia, y existe microcitosis importante e hipocromía leve.
En estado homocigoto (o doble heterocigoto), la talasemia es intermedia o mayor, estos pacientes tienen importante anemia microcítica hipocrómica y reticulocitosis, (aunque no la esperable para el grado de hemólisis), además, existe hepatomegalia, esplenomegalia y deformación de los huesos.
Las talasemias β (beta) se caracterizan por deficiencia cuantitativa de cadenas de globina β subyacente a una sorprendente heterogeneidad de defectos moleculares.
Las mutaciones que inactivan por completo el gen β y no producen globina β se denominan β 0 (Beta “cero”). Otras mutaciones permiten la producción de algo de globina β y, según el grado de reducción cuantitativa en la producción de las cadenas β, se clasifican como talasemia β + - o β ++ - (“silenciosa”).
Una reducción cuantitativa en la globina β da como resultado la acumulación de cadenas de globina alfa excedentes que precipitan en los eritrocitos, produciendo hemólisis y también lo hacen en los eritroblastos medulares produciendo hematopoyesis ineficaz.
La enfermedad de células falciformes es un término general que define un grupo de enfermedades hereditarias (que incluyen anemia de células falciformes o hemoglobinopatía SS, la hemoglobinopatía SC y la hemoglobinopatía Sβ-talasemia) caracterizadas por mutaciones cualitativas en el gen que codifica para la cadena globínica β, uno de los componentes de la hemoglobina.
Esta hemoglobina se llama S (Slow) porque electroforéticamente migra más lento que la hemoglobina A.
El defecto cualitativo está en el sexto aminoácido, ya que en lugar de haber un ácido glutámico en esta posición hay una valina.
Este cambio produce una modificación conformacional en la estructura tridimensional de la molécula de la hemoglobina que hace que en condiciones de desoxigenación (es decir, cuando la Hb no está unida al oxígeno), los tetrámeros de Hb -que incluyen dos de estas subunidades mutantes de globina β falciforme-, es decir, HbS, se polimericen formando largos cristales dentro de los glóbulos rojos adoptando estas células una forma de media luna u hoz, de donde la enfermedad toma su nombre.
Los drepanocitos (células falciformes) dan origen a dos problemas importantes en los homocigotos para hemoglobina S: se destruyen prematuramente (dando origen a una grave anemia hemolítica) y producen cuadros de oclusión vascular en varios órganos, lo que lleva a crisis de dolor importante, hipoxia y frecuentemente infartos óseos, cerebrales, pulmonares, esplénicos, renales.
La incidencia poblacional es aproximadamente del 1%; afortunadamente la mayoría de estos pacientes son heterocigotos asintomáticos, teniendo incluso alguna ventaja pues están parcialmente protegidos de la infección con Plasmodium falciparum.
La Hb A, es la forma de hemoglobina más abundante (>90%) en la vida adulta, está formada por dos subunidades de globina alfa (codificadas por el cromosoma 16) y dos subunidades de globina beta (cromosoma 11). La sustitución de un solo nucleótido en la globina β da como resultado el alelo β S.
Los tetrámeros de Hb con una sóla globina βS (estado heterocigoto) también pueden polimerizarse, aunque no tan eficientemente como la HbSS, lo que hace que los sujetos heterocigotos sean generalmente asintomáticos; sólo cuando se exponen a presiones parciales de oxígeno bajas pueden formar drepanocitos y tener crisis de oclusión vascular.
Son posibles otros genotipos de la enfermedad de células falciformes. En la hemoglobinopatía S-beta talasemia, el alelo βS se combina con un alelo β cero talasémico (Hbβ0), por lo que no se producen cadenas beta normales y predominan las cadenas βS, que además de ser anormales y polimerizar, son pocas. Por lo tanto en esta hemoglobinopatía talasémica se encuentra microcitosis, hipocromía y reticulocitosis, además de drepanocitos, frecuentemente hipocrómicos.
El alelo βS combinado con un alelo β + talasémico (Hbβ+), es decir, que producen un poco más que cero, da como resultado la hemoglobinopatía-talasémica HbSβ+ , enfermedad generalmente más leve que HbSβ0 debido a la expresión de cuando menos algo de HbA normal.
El objetivo del artículo es presentar el abordaje y la integración de herramientas diagnósticas, desde las más sencillas hasta el uso equipos completamente automatizados (MINICAP Flex Piercing Sebia), de una familia mexicana, con una clásica combinación de mutaciones hereditarias leves en las globinas beta que se combinaron para ocasionar un cuadro más grave.
Presentación del Caso Clínico
Masculino (Papá) de 40 años de edad, completamente asintomático. La biometría hemática fue completamente normal (imagen 1). Los frotis de sangre periférica (fotomicrografía 1A) no mostraron ninguna alteración morfológica. Debido a los hallazgos encontrados en la hija (ver abajo) se realizó una inducción de drepanocitos que fue positiva encontrándose drepanocitos cortos, frecuentes en los sujetos heterocigotos para hemoglobina S (fotomicrografía 1B).



La electroforesis de hemoglobina (imagen 2) mostró la presencia de un pico en la zona S (39.4%) y disminución porcentual de HbA (57.6%). La HbA2 fue normal. El diagnóstico final fue heterocigoto para hemoglobina S.
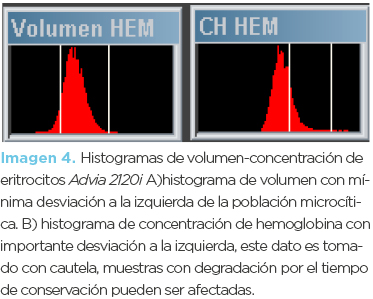
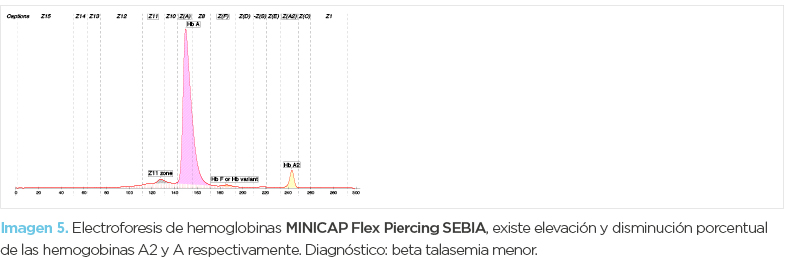
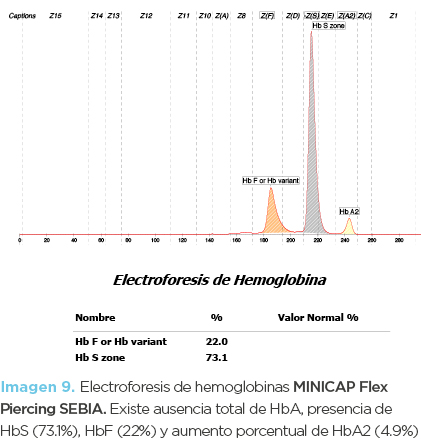
Femenino (Mamá) de 45 años de edad. El informe de serie roja (imagen 3) mostró anemia microcítica-normocrómica así como mínima reticulocitosis; en este caso no se encontró aumento de glóbulos rojos. Los histogramas de serie roja volumen/concentración (imagen 4) (Advia 2120i) mostraron poblaciones microcíticas hipocrómicas. En los frotis (fotomicrografía 2) se observaron eritrocitos microcíticos hipocrómicos, moderada cantidad de codocitos, poiquilocitos, y eritrocitos con punteado basófilo, la inducción de drepanocitos fue negativa. La electroforesis de hemoglobina (imagen 5) mostró incremento porcentual de HbA2 (5.4%), disminución de HbA1 (92.1%) y sutil expresión de HbF (hemoglobina fetal). El diagnóstico final fue beta-talasemia menor.


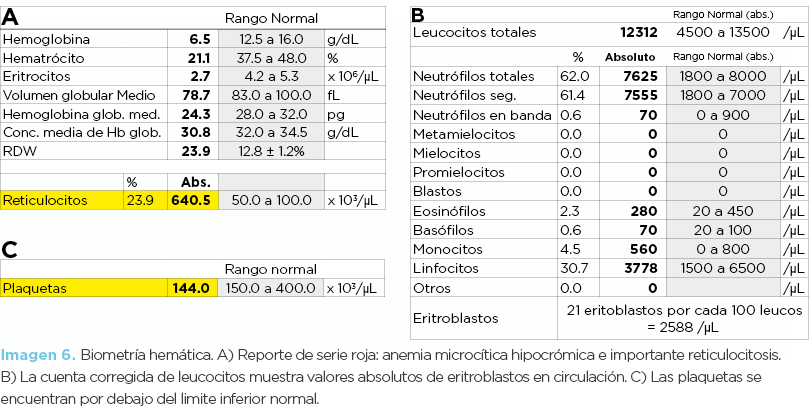
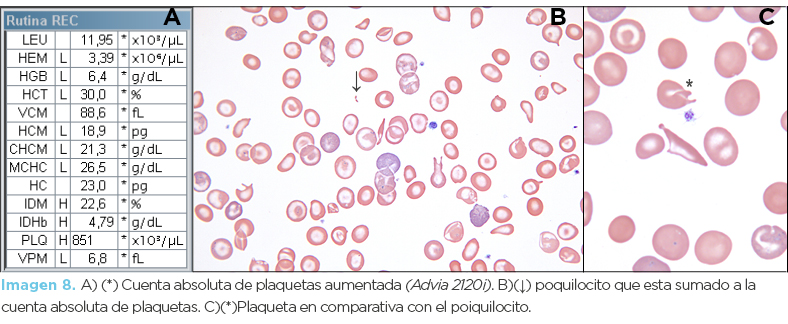
Niña de 10 años de edad (hija menor; paciente propositus). La biometría hemática (imagen 6 y 7) mostró importante anemia microcítica hipocrómica, incremento de reticulocitos y presencia de eritroblastos. Además se encontró mínima trombocitopenia. Curiosamente en el equipo Advia 2120i se encontró “trombocitosis”; la razón de esto es porque existen abundantes poiquilocitos que por su tamaño el aparato confunde con plaquetas (imagen 8). Los frotis muestran abundantes codocitos, notable cantidad de reticulocitos de stress y eritroblastos, así como algunos drepanocitos hipocrómicos o hipercrómicos. La inducción de drepanocitos fue positiva (fotomicrografía 3).
La electroforesis de hemoglobinas en mi mostró ausencia total de HbA, presencia de HbS (73.1%), HbF (22%) y aumento porcentual de HbA2 (4.9%) (imagen 9). El diagnóstico final fue hemoglobinopatía S-beta talasemia.


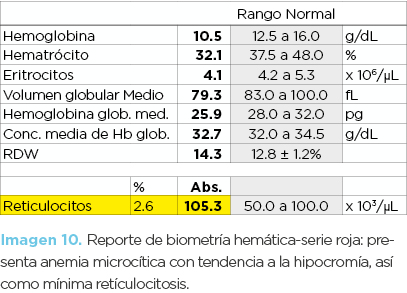
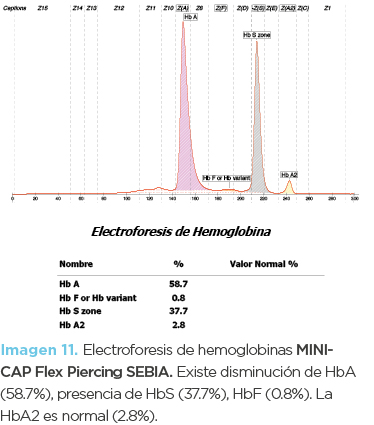
Niña de 11 años de edad (hija mayor). La biometría hemática mostró anemia microcítica-normocrómica, con mínima reticulocitosis (imagen 10). En los frotis no se observaron alteraciones significativas. La inducción de drepanocitos mostró drepanocitos cortos (similares a los de la fotomicrografía 1) y la electroforesis de hemoglobinas disminución de HbA (58.7%), presencia de HbS (37.7%) y HbF (0.8 %). La Hb A2 fue normal (imagen 11). El diagnóstico fue de hemoglobinopatía S heterocigota asociada a posible deficiencia de hierro.
Conclusión
El abordaje de los defectos cualitativos y cuantitativos de las cadenas de beta globina, en el caso particular de esta familia, se inició con la historia clínica de la paciente propositus, la hija menor, quien desde el nacimiento habían presentado importante anemia microcítica-hipocrómica.
Para un diagnóstico preciso de la etiología en casos de anemia se debe de partir de la clasificación morfológica de dicha anemia (microcítica hipocrómica, normocítica normocrómica, macrocítica normocrómica, etc.), de la observación del frotis y complementar con los estudios necesarios que en este tipo de casos incluyen la inducción de drepanocitos, la electroforesis de hemoglobinas, el perfil de hierro y el estudio familiar (cuadro 1).
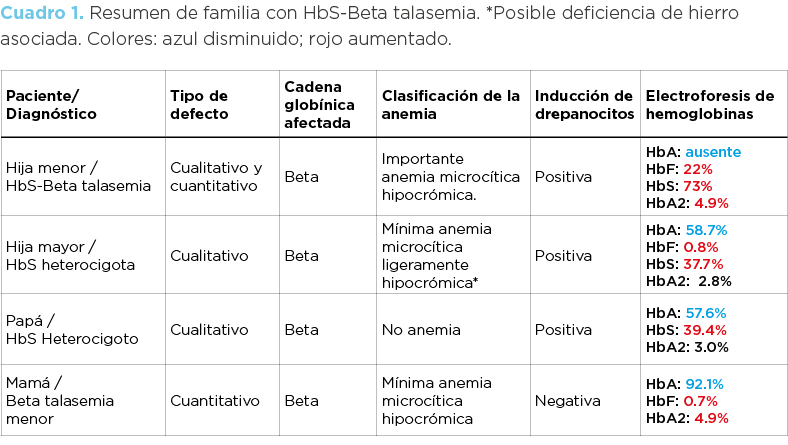
La Hemoglobinopatía S-Beta talasemia de la paciente tiene dos componentes.
Cuantitativo: La disminución de síntesis de globina beta (la paciente tiene un gen beta cero) ocasiona anemia microcítica hipocrómica; además, al existir deficiencia de cadenas beta, se produce un exceso relativo de cadenas alfa que se precipitan en los eritroblastos medulares, dando lugar a eritropoyesis ineficaz y en los eritrocitos, dando lugar a hemólisis y aumento de reticulocitos; ambos fenómenos incrementan la gravedad de la anemia, además los precipitados de las cadenas alfa excedentes son removidos cuando los eritroblastos salen del espacio medular a la sangre o cuando los eritrocitos pasan de los cordones de Billroth a los sinusoides esplénicos; la remoción de los precipitados da lugar a los poiquilocitos.
Cualitativo: La cadena de la beta globina S (la paciente tiene un gen beta S) se produce en cantidad normal pero se polimeriza cuando está desoxigenada provocando la formación de drepanocitos, que incrementan la hemólisis.
En la electroforesis de hemoglobinas de la paciente propositus hay ausencia total de la HbA porque no hay ninguna cadena beta normal: uno de los genes es β0 y el otro βS. En estos casos, frecuentemente está elevada la hemoglobina fetal, probablemente por un aumento compensatorio de síntesis de cadenas gamma (la hemoglobina fetal está compuesta por dos cadenas alfa y dos gamma). Por otra parte la hemoglobina A2 (dos cadenas alfa y dos delta) está elevada porcentualmente, no por una elevación real sino por la disminución de hemoglobina A con aumento relativo de de la A2, como sucede en las beta-talasemias menores (ejemplo, la madre).
Esta combinación, anemia microcítica hipocrómica con reticulocitos elevados, presencia de drepanocitos, electroforesis de hemoglobina con ausencia de A1, presencia de hemoglobina S y aumento porcentual de A2 es típico de la hemoglobinopatía S-beta talasemia.
La mayoría de estas enfermedades hereditarias no tiene un tratamiento específico, por lo que, la importancia de establecer el diagnóstico radica en el consejo genético para futuras generaciones.
El papá y la mamá tienen cuadros clásicos de HbS heterocigoto y beta-talasemia menor respectivamente; sin embargo la hija mayor, que es heterocigota para hemoglobina S, presenta mínima anemia microcítica hipocrómica, que probablemente está asociada a deficiencia de hierro.