Introducción
La enfermedad de von Willebrand (EvW) es el trastorno hemorrágico hereditario más común en los seres humanos. El trastorno muestra una distribución mundial y también es común en otras especies (animales, incluidos perros y cerdos), se define por la disminución de la actividad del factor von Willebrand (FvW) en la sangre. El FvW es una glicoproteína multimérica sintetizada en células endoteliales y megacariocitos y luego almacenada dentro de los cuerpos de Weibel Palade y los gránulos alfa, respectivamente. Se elimina mediante macrófagos en el hígado y el bazo.
Es un trastorno hereditario heterogéneo, tanto clínica como genéticamente, causado por alteraciones funcionales y/o cuantitativas de la proteína. La característica central de todos los tipos de EvW es la presencia de cantidades reducidas del FvW o de formas anormales del mismo en el torrente sanguíneo. Esto puede ser debido a un defecto cuantitativo o cualitativo. La falta de función del FvW, conduce a un fenotipo hemorrágico debido a su papel fundamental en la hemostasia primaria y secundaria. Dentro de la hemostasia primaria, el FvW se une a proteínas de la matriz extracelular como el colágeno, así como a las plaquetas a través del receptor de la glicoproteína Ib. En la hemostasia secundaria, el FvW circula unido al factor VIII (FVIII) de la coagulación protegiéndolo de la degradación por proteasas plasmáticas.
El diagnóstico y la subclasificación de la EvW siguen planteando importantes desafíos clínicos. Esto se puede atribuir, en parte, al hecho de que los niveles plasmáticos del FvW varían en un amplio rango en la población normal, junto con las múltiples funciones fisiológicas que desempeña in vivo. En los últimos años, se han logrado avances sustanciales en el esclarecimiento de las funciones biológicas del FvW. También se han logrado avances significativos en la definición de los mecanismos fisiopatológicos que sustentan la EvW tanto cuantitativa como cualitativa. En particular, se han desarrollado varios ensayos de laboratorio nuevos que permiten una evaluación más precisa de aspectos específicos de la actividad del FvW.
Historia
La primera descripción la realizó Erik Adolf von Willebrand, en 1926, quien inicialmente consideró un problema de sangrado mucocutáneo severo presente en un pedigrí familiar de 58 individuos de dos familias interrelacionadas que abarcaban cuatro generaciones. El probando era una niña de cinco años que presentó sangrado mucoso severo recurrente y en las pruebas demostró un tiempo de coagulación y retracción del coágulo normales, pero una marcada prolongación de su tiempo de sangrado. Dentro del pedigrí, tanto hombres como mujeres se vieron afectados, y el sangrado fue severo, con múltiples miembros femeninos de la familia, incluida la niña, que murieron como consecuencia del sangrado de las mucosas. (1)
Las pruebas de diagnóstico disponibles desde principios hasta mediados del siglo XX eran inespecíficas, engorrosas, no reproducibles y requerían mucho tiempo. La medición de la actividad de coagulación del FVIII (FVIII:C) estuvo disponible por primera vez a mediados del siglo XX y generó confusión en el diagnóstico, ya que se observó una deficiencia en la hemofilia clásica, así como en la afección definida posteriormente como EvW. Esto llevó a la etiqueta de “pseudohemofilia”, aunque otros llamaron al trastorno “hemofilia vascular”, ya que se planteó la hipótesis de que el tiempo de sangrado prolongado podría ser secundario a defectos capilares.
Durante los años cincuenta se descubrió una relación con la disminución de la actividad del FVIII, que lograba ser corregida con plasma de pacientes sanos. Durante la década de los sesenta, el noruego Christian Borchgrevink vislumbró los primeros indicios del rol del FvW en la agregación plaquetaria, información confirmada en 1970 por Margaret Howard y Barry Firkin a través de sus estudios con ristocetina, un antibiótico que induce el proceso de agregación en individuos sanos y hemofílicos, pero no en pacientes con EvW. Finalmente, hacia 1985 el gen del FvW es aislado y en 1989 se determina su estructura, gracias a lo cual se confirma que la EvW y la hemofilia A son entidades distintas codificadas por genes independientes (2).
VWD: Clasificación y características clínicas
Se clasifica en: Alteraciones cuantitativas Tipo 1 (deficiencia parcial) y Tipo 3 (deficiencia total) y defectos cualitativos (Tipos: 2A, 2M, 2B y 2N).
Clínicamente, los pacientes con EvW experimentan sangrado mucocutáneo excesivo, que incluye sangrado menstrual abundante, epistaxis, fácil formación de hematomas, sangrado prolongado de heridas menores y de la cavidad bucal, y sangrado gastrointestinal, así como sangrados secundarios a procedimientos odontológicos, partos y cirugías con sangrado musculoesquelético (vistos en casos graves).
Diagnóstico
La prevalencia de la EvW varía de 1 en100 a 1 en 10,000 personas (4). Debido a que el FvW es una glicoproteína multimérica y multifuncional con varios dominios que contienen diversos sitios de unión, se requiere más de una prueba y varios pasos para evaluar todas sus funciones, los cuales serían:
1) la evaluación del historial de sangrado personal y familiar del paciente,
2) detección inicial de trastornos hemorrágicos, 3) pruebas para la detección de la EvW, 4) pruebas para la tipificación de EvW y, por último, 5) el análisis molecular.
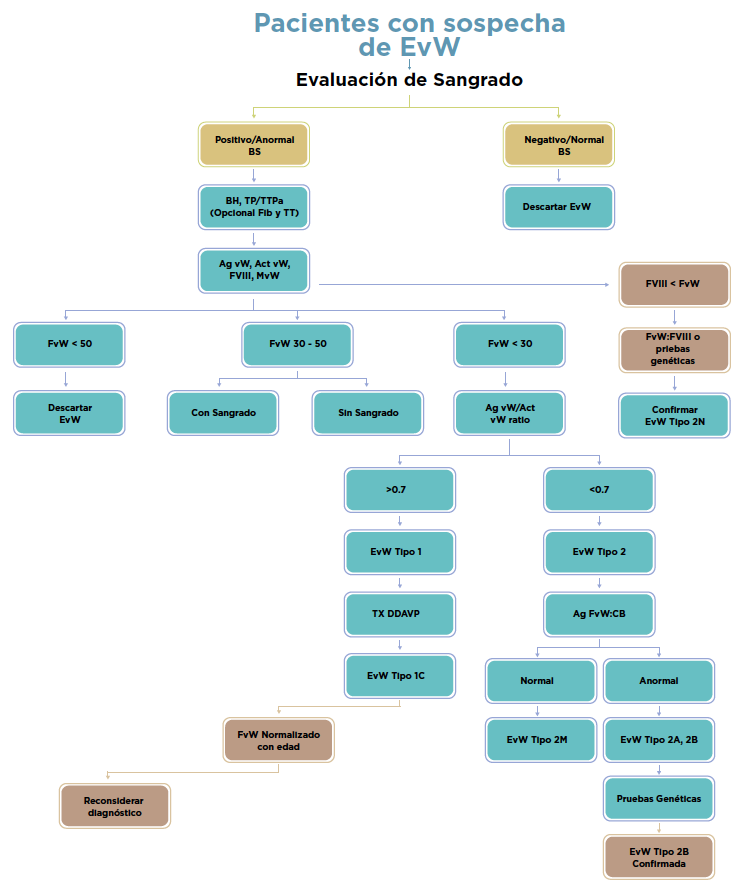
En la guía diagnóstica basada en evidencia, consensuada entre 4 sociedades científicas: Sociedad Americana de Hematología, Sociedad Internacional de Trombosis y Hemostasia, Fundación Nacional de la Hemofilia de Estados Unidos y la Federación Mundial de la Hemofilia, se presentan las recomendaciones y los pasos a seguir para el diagnóstico de los diferentes tipos de EvW. Así como un algoritmo general para el diagnóstico de la EvW. (Ver figura 1)
rombocitopenia leve puede ocurrir en pacientes con tipo 2B. El tiempo de hemorragia (TH) usualmente está prolongado, pero puede ser normal en pacientes con formas leves de la enfermedad, como ocurre en el tipo 1. El tiempo de protrombina (TP) es normal y el tiempo de tromboplastina parcial activado (TTPa) puede estar prolongado de acuerdo con la concentración del FVIII. El FvW: Antigénico (FvW: Ag) y el cofactor de ristocetina (FvW: RCo) son las pruebas básicas para establecer el diagnóstico de EvW. Estudios adicionales, como la agregación plaquetaria inducida por ristocetina (RIPA) y el estudio de los multímeros, permiten caracterizar a la enfermedad de von Willebrand para un tratamiento apropiado. (5)
La evaluación de multímeros del factor de von Willebrand (MFvW) es parte esencial del diagnóstico de algunas variantes de EvW. Mediante técnicas electroforéticas adecuadas se pueden estimar los multímeros de peso molecular bajo (LMWM), intermedio (IMWM), alto (HMWM) y ultra-alto (UHMWM). (6) Sin embargo se trata de una de las metodologías más laboriosas, con tiempos de proceso largos y con poca estandarización. Además de que exige amplia experiencia por lo que su uso se reduce a centros altamente especializados en el estudio de la EvW, de investigación o laboratorios de referencia.
La aparición de un kit comercial semiautomatizado plantea una alternativa para que más laboratorios de Hemostasia puedan llevar a cabo el ensayo, como fue demostrado en el artículo “Evaluación preclínica de un ensayo de electroforesis comercial rápido y semiautomático para multímeros del factor von Willebrand”, el cuál menciona lo siguiente: el ensayo de MFvW de Sebia es fácil de realizar y puede implementarse con éxito en cualquier laboratorio clínico para la evaluación de segunda etapa de la EvW. El poder de resolución de la distribución de multímeros es adecuado para clasificar correctamente la EvW tipos 1, 2A, 2B y 3 (7). Además de reducir los tiempos de proceso a menos de 6 horas.
Conclusión
Es un hecho demostrado que la EvW es el trastorno hemorrágico con mayor prevalencia a nivel mundial, es indispensable realizar un adecuado diagnóstico para poder clasificarla y también, diferenciarla de una hemofilia realizando las distintas pruebas de tamizaje (cuadro hemático, adhesividad plaquetaria, tiempos de coagulación), especiales (FvW:Ag, Actividad del FvW, FVIII:C) y diagnósticas (multímeros, RIPA, FvW:CB, FvW:FVIIIB y test de desmopresina), así como el abordaje del algoritmo diagnóstico establecido en guías internacionales.
Debido a que las pruebas genéticas no están al alcance de todos los pacientes, la llegada del Kit hydragel (5 y 11), ha revolucionado la determinación multimérica del FvW y el diagnóstico acertado de esta según la clasificación actual : Tipo 1 (1, 1C), Tipo 2 (A,B,N,M) y Tipo 3.
Bibliografía:
1. Weyand, A. C., & Flood, V. H. (2021). Von Willebrand Disease: Current Status of Diagnosis and Management. Hematology/oncology clinics of North America, 35(6), 1085–1101. https://doi.org/10.1016/j.hoc.2021.07.004
2. Tovar Sánchez, C. V., Salazar Reviakina, A., Rumbo Romero, J. A., Sierra Bretón, M. M., Madariaga Perpiñán, I., & Zarante Montoya, I. M. (2020). ¿Qué avances recientes hay en el entendimiento, diagnóstico y tratamiento de la enfermedad de Von Willebrand?: una revisión de la literatura. Universitas Médica, 61(2). https://doi.org/10.11144/javeriana.umed61-2.vonw
3. James, P. D., Connell, N. T., Ameer, B., Di Paola, J., Eikenboom, J., Giraud, N., Haberichter, S., Jacobs-Pratt, V., Konkle, B., McLintock, C., McRae, S., R. Montgomery, R., O’Donnell, J. S., Scappe, N., Sidonio, R., Jr, Flood, V. H., Husainat, N., Kalot, M. A., & Mustafa, R. A. (2021). ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Advances, 5(1), 280–300. https://doi.org/10.1182/bloodadvances.2020003265
4. Mejía-Buriticá, L., Pérez-Monterrosa, M. E., & Vizcaíno-Carruyo, J. C. (2023). Diagnóstico de la enfermedad de von Willebrand. Medicina Y Laboratorio, 27(2), 139–155. Recuperado a partir de https://medicinaylaboratorio.com/index.php/myl/article/view/634
5. Martínez-Murillo, C. (2018). The challenge in the diagnosis and treatment. Rev Hematol Mex, 19(2), 61–62.
6. Lopez, M. S., Paiva, J., Woods, A., Saez, M. S., Barrera, L. H., Privitera, V., Chuliber, F., Villagra Iturre, M., Penchasky, D., Sorroche, P., Martinuzzo, M., & Sánchez Luceros, A. (2022). Comparación de perfiles multiméricos del factor de von Willebrand obtenidos mediante un ensayo electroforético comercial y electroforesis local con geles de agarosa 1%. Revista Hematología, 26(2), 9–19. https://doi.org/10.48057/hematologa.v26i2.441
7. Pikta, M., Zemtsovskaja, G., Bautista, H., Nouadje, G., Szanto, T., Viigimaa, M., & Banys, V. (2018). Preclinical evaluation of a semi-automated and rapid commercial electrophoresis assay for von Willebrand factor multimers. Journal of clinical laboratory analysis, 32(6), e22416. https://doi.org/10.1002/jcla.22416