LA PLAQUETA Y SU ESTRUCTURA.
Las plaquetas son células anucleadas de 1–2 μm de tamaño, generadas en la médula ósea por fragmentación de los bordes de los megacariocitos. El intervalo fisiológico de las plaquetas es de 150–400 × 109/L. Un adulto sano produce cada día una media de alrededor de 1×1011 plaquetas. La expectativa de vida de las plaquetas es de 7 a 10 días.
Las plaquetas tienen características muy particulares que les permiten una función activa y crucial cuando hay daño en los vasos. Su membrana les confiere una gran comunicación con el medio que las rodea, no sólo para recibir estímulos sino para responder en consecuencia, una vez activadas, se generan reacciones intracelulares con efecto sobre las proteínas contráctiles que retraen a las plaquetas y favorecen la degranulación, con lo que se incrementan las concentraciones de sustancias activas para los procesos de agregación y coagulación.
Gránulos y organelos de las plaquetas.
Las plaquetas logran su función gracias al efecto amplificador que se obtiene con el estímulo que reciben, por lo que la liberación del contenido de sus gránulos es indispensable para sostener la activación hasta el final. Tienen dos tipos de gránulos, los a y los densos; además, poseen microperoxisomas, vesículas, mitocondrias y depósitos de glucógeno.
Gránulos a. Cada plaqueta contiene alrededor de 50 gránulos, de aproximadamente 300 nm de diámetro dentro del contenido de estos gránulos encontramos moléculas como b-tromboglobulina, factor 4 plaquetario y trombospondina, Factor de von Willebrand (FvW) y factor V, solo por mencionar algunos.
Gránulos densos.
Tienen alrededor de 250 nm de diámetro y se pueden encontrar aproximadamente cinco por plaqueta, constituyen un reservorio principalmente de ADP, Ca2+, ATP, pirofosfato y serotonina que se incorpora desde el plasma.
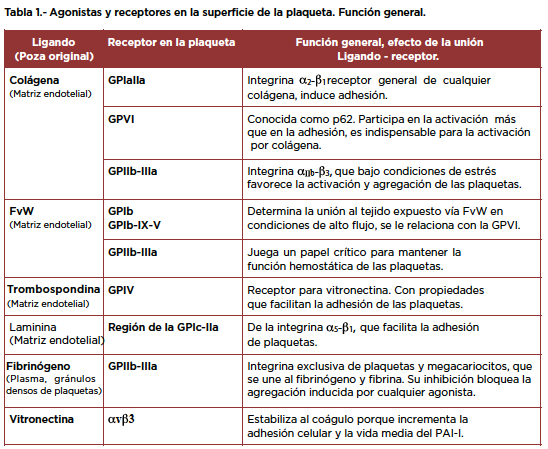
En la tabla 1 se exponen las Glicoproteínas de membrana (GP) más importantes, sus ligandos en el exterior y los efectos que inducen al unirse a las plaquetas.
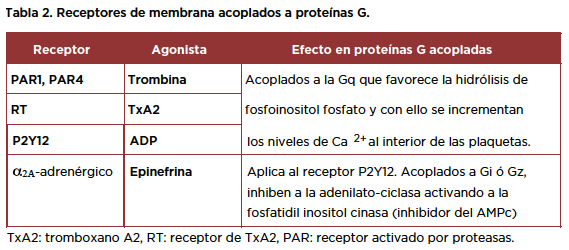
Receptores acoplados a proteínas G.
La mayor parte de las reacciones intracelulares se lleva a cabo gracias a la activación de proteínas G acopladas a receptores de membrana, cuya estructura general, incluye una secuencia aminoacídica con una fracción extracelular N-terminal que les permite ser reconocidos por diferentes agonistas.
Como se menciona en la tabla 2.
FUNCIÓN DE LAS PLAQUETAS EN LA HEMOSTASIA
La presencia de las plaquetas sanas y en cantidades adecuadas, es indispensable para la hemostasia normal. De ellas depende la localización y retracción del coágulo en el sitio de lesión, gracias a que se adhieren y agregan específicamente en el área donde se exponen la colágena, el factor de von Willebrand y el factor tisular (FT).
En la superficie de las plaquetas se forma la trombina responsable de la estabilización del coágulo hemostático, puesto que participa en la polimerización de la fibrina, activa al factor XIII, al TAFI, rompe los receptores PAR-1 sobre la plaqueta y se incluye en el coágulo para estabilizarlo.
Activación de las plaquetas:
El proceso se inicia en el sitio de lesión, la colágena expuesta induce la adhesión de las plaquetas, ya sea de forma directa a través de los receptores GPVI ó de forma indirecta por el FvW; debido a que la mayoría de las plaquetas circulan a alta velocidad, no hay adhesión masiva al sitio de lesión, son las plaquetas “rodantes” sobre el endotelio las que se unen al tejido expuesto y se activan. En condiciones de bajo flujo, los receptores primarios de la colágena son GP Ia-IIa, VI y IV, mientras que, en condiciones de alto flujo, la interacción es vía GP Ib y GP IIb-IIIa, que en condiciones de bajo flujo se une mejor al fibrinógeno.
Gracias a la secreción de los gránulos de las primeras plaquetas, se generan concentraciones de trombina, ADP y TxA2 suficientes para reclutar más plaquetas. El estudio de la participación celular en la formación del coágulo ha propuesto la existencia de diferentes subpoblaciones de plaquetas activadas: tal es el caso de las plaquetas COAT (plaquetas estimuladas por colágena y Trombina), que durante la exposición inicial de la colágena y la generación de las primeras trazas de trombina, induce en las plaquetas adheridas a la matriz expuesta una mayor activación que la que se genera en las plaquetas reclutadas cuando van cubriendo espacios en el tapón plaquetario.
Se ha demostrado que la activación conjunta por colágena y trombina, favorece la unión de mayor cantidad de factor V proveniente de los gránulos de la plaqueta, y de los factores VIII, IX y X; al mismo tiempo, la explosión en la generación de factor Xa y trombina correlaciona con la cantidad de factores de la coagulación unidos a los fosfolipidos (FL) de la plaqueta y no exclusivamente con la concentración de FL expuestos, lo que depende en gran medida de la activación de la GPVI, circunstancia que no se produce cuando sólo se activa por alguno de los agonistas de forma independiente, de esta manera, las plaquetas que se reclutan sobre la monocapa inicial no reciben el estímulo conjunto de la colágena y trombina, limitando su respuesta en la unión de factores y generación de factor Xa y trombina. No obstante, el estímulo permite que las plaquetas activadas emitan pseudópodos gracias al reordenamiento del citoesqueleto para amplificar la superficie de reacción.
La activación más eficiente incluye tanto a las proteínas Gq, como a las Gi, de forma que se favorece la liberación del calcio intra-citoplasmático y se inhibe la síntesis del AMPc proveniente de las células endoteliales. Una vez activadas, el sistema pasa a la fase de propagación, donde se generan grandes cantidades de trombina; las plaquetas reclutadas, se activan y soportan vía fosfatidilserina (FS) los componentes necesarios para la formación del coágulo estable, al mismo tiempo, se generan señales hacia el interior y hacia el exterior de las plaquetas para favorecer la retracción del coágulo y su estabilización, gracias a la participación de diferentes moléculas en la superficie de la membrana plaquetaria.
En estado pasivo las plaquetas no ponen en contacto tales moléculas; sin embargo, una vez que se han activado, la agregación primaria de las plaquetas permite que los ligandos estén en contacto el tiempo suficiente y a la distancia necesaria para que se reconozcan y desencadenen señales intracelulares para la reorganización de su citoesqueleto, secreción de los gránulos a y adhesión al fibrinógeno, dado que se modifica una porción extracelular del receptor del fibrinógeno GPIIb-IIIa, se cambia su estado de baja afinidad a un estado de alta afinidad, lo que facilita la agregación irreversible de las plaquetas (agregación secundaria) y posterior retracción del coágulo.
En la retracción del coágulo, el calcio iónico liberado, activa a la miosina-cinasa que fosforila su cadena ligera, la miosina fosforilada, ahora de alta afinidad por la actina, se asocia a esta última, formando la “actomiosina”, que induce la contracción del citoesqueleto de la plaqueta que se encuentra anclado de manera intra-membrana a los receptores GpIIb, IIIa del fibrinógeno (ahora fibrina), facilitando la retracción del tapón plaquetario de manera que el coágulo formado, insoluble por la acción del fXIII sobre la fibrina, es capaz de resistir el flujo de la sangre que circula hasta el momento de la lisis total de ese “tapón”.
La participación de las plaquetas en la formación del tapón plaquetario, es entonces, el fin primordial de su existencia y tiene tantas repercusiones, que se ha propuesto una ampliación al modelo celular, relacionada al aporte que recibe el proceso de hemostasia vía agonistas derivados de los gránulos plaquetarios sobre el factor VIIa en experimentos in vitro, independientemente del factor tisular.
Las alteraciones en la plaqueta pueden conducir a graves problemas en el proceso hemostático del paciente, tanto trombóticos como hemorrágicos.
Los principales tipos de alteraciones de plaquetas son:
La trombocitemia: Es la sobreproducción de plaquetas en la sangre, causada por una mutación genética o por factores externos como la inflamación.
Trombocitopenia: Disminución del número de plaquetas que puede producirse a raíz de una alteración de la medula ósea, como la leucemia o un problema del sistema inmunitario, o bien, puede ser efecto secundario a ciertos medicamentos o infecciones.
Disfunción plaquetaria: En este caso, los niveles de plaquetas pueden ser normales, pero existe un problema con el funcionamiento. A menudo son situaciones adquiridas ya que las plaquetas son sensibles a muchos medicamentos e incluso a la ingesta de ciertos alimentos, aunque también se puede tratar de enfermedades hereditarias.
Las pruebas de laboratorio dirigidas al estudio de las plaquetas se clasifican en 2 tipos: las que estudian la cantidad y las que evalúan la función. Entre las que evalúan la función se encuentra la agregación plaquetaria; esta prueba comenzó a utilizarse en 1962 cuando Born la describió y explicó cómo es que se agregan las plaquetas. La inducción de la agregación plaquetaria in vitro es un fenómeno que se estudia en el laboratorio clínico utilizando sustancias con capacidad agregante agonista como adenosín difosfato (ADP), epinefrina, colágeno, TRAP-6 (agonistas) y ristocetina.
Conclusión
En resumen, las plaquetas juegan un papel fundamental en la hemostasia y la reparación de tejidos. Su capacidad para adherirse, activarse y formar coágulos asegura la detención del sangrado y la restauración de la integridad vascular. El estudio de la función de las plaquetas por las pruebas de agregación plaquetaria son fundamentales para comprender los mecanismos subyacentes a trastornos hemorrágicos y trombóticos. Estas pruebas proporcionan información valiosa sobre la capacidad de las plaquetas para formar coágulos y su respuesta a diferentes estímulos. Además, permiten la evaluación de la eficacia de tratamientos antiplaquetarios.
Referencias:
1.- Mellón LA. Fisiología de la Hemostasia en Fisiología Humana 2ª. edición. Ed. Mxgraw-Hill Interamericana Tresguerres JAF.
2.- Moroi M, Jung S M: Platelet Receptors for collagen. Thrombosis and Haemostasis 1997; 78(1):439-444.
3.- Kehrel, B, Wierwille S, Clemetson KJ, Anders O, Steiner M, Knight G, Farndale RW, Okuma M, Barnes MJ: Glycoprotein VI is a mayor collagen receptor for platelet activation: it recognizes platelet-activating quaternary structure of collagen, whereas CD36, glycoprotein IIb-IIIa, and von Willebrand Factor do not. Blood 1998; 91(2):491-499.
4.- Kleinschnitz V, Pozgajova M, PhamM, Bendszus M, NieswandtB, StoolG: Targeting platelets in acute experimental stroke: Impact of glycoprotein Ib,VI, and IIb/IIIablockade on infarct size, functional otucome, and intracranial bleeding. Circulation 2007; 115;2323-2330.
5.- Brass LF, Molino M: Protease-activated G protein-coupled receptors on human platelets and endotelial cells. Thrombosis and Haemostasis 1997; 78(1):234-241.
6.- Brass LF: Thrombin and platelet activation. CHEST 2003; 124:18S-25S.
Spronk MH, van der Voort D, Ten CH: Blood coagulation and the risk of atherothrombosis: a complex relationship. Review. Tthrombosis Jorunal 2004; 2:12.
7.- Jamieson GA: Pathophysiology of platelet thrombin receptors. Thrombosis and haemostasis 1997; 78(1):242-246.
Hoffman M, Monroe D: Coagulation 2006: A modern view of hemostasis. Hematol Oncol Clin N Ann 2007;211:1-11.
8.- Kempton L, Hoffman M, Roberts H, Monroe D: Platelet heterogeneity, variation in coagulation complexes on platelet subpopulations. Arterioscler Thromb Vasc Biol 2005; 25:861-866.
9.- Ilveskero S, Siljander P, Lassila R: Procoagulant activity on platelets adhered to collagen or plasma clot. Arter Thromb and Vasc Biol 2001; 21:628-635.
10.- Keuren J, Wielders S, Ulrichts H, Hackeng T, Heemskerk J, Deckmyn H, et al: Synergistic effect of thrombin on collagen-induced platelet procoagulant activity is mediated through protease-activated receptor-1. Arterioscler Thromb Vasc Biol 2005; 25:1499-1505.
11.- Heemskerk JWN, Bevers EM: Platelet activation and blood coagulation. Thromb Haemost 2002; 88:186-193.
12.- Prevost N, Woulfe DS, Tognolini M, Tanaka T, Jian W, Frota RR: Signaling byephrin B1 and Eph kinasas in platelets promotes Rap1 activation, platelet adhesión, and aggregation via effector pahtways that do not require phosphorylation of ephrin B1. Hemos Thromb Vasc Biol 2004; 103:1348-1355.
Prevost N, Woulfe DS, Tanaka T, Brass LF: Interactions between Eph kinases and ephrins provide a mechanism to support platelet aggregation once cell-to-cell contact has ocurred. PNAS 2002; 99(14):9219-9224.
13.- Altman R, Scazziota AS, Herrera ML González C: Thrombin generation by activated factor VII on platelert activated by different agonists. Extending the cell-based model of hemostasis. Thrombosis Journal 2006; 4(5):1-8.