Infraestructura y equipamiento del laboratorio
La calidad de los resultados de las pruebas que proporciona un laboratorio de virología clínica depende en gran medida de sus instalaciones, configuración organizativa y equipo utilizado. En general, el laboratorio estará organizado en áreas funcionales diseñadas para proporcionar un servicio seguro y eficaz. Entorno que garantice la prestación de los servicios de diagnóstico con los estándares regulatorios y de calidad adecuados.
Por ejemplo, la mayoría de los laboratorios de virología clínica tendrán áreas funcionales separadas según las siguientes líneas:
• Zona de recolección y preparación de muestras.
• Área general de laboratorio, almacenes, sala de máquinas, etc.
• Virología molecular.
• Serología.
•Unidad de cultivo y aislamiento de virus (Nota: en algunos laboratorios especializados en virología esto podría dividirse aún más dependiendo del tipo y nivel de riesgo del patógeno viral).
• Zona de oficinas fuera del laboratorio.
Cuatro áreas o salas designadas brindan al laboratorio la capacidad de separar las actividades de manera eficiente, como son: área de preparación de reactivo, el área de extracción/procesamiento de muestras, la sala de configuración del ensayo (etapa de adición de plantilla) y el área de amplificación molecular.
Cada sala dispone de su propio equipamiento como campanas de seguridad biológica, pipetas, centrífugas, refrigerador y congelador, además de un almacén exclusivo de consumibles/reactivos cuyo uso está restringido dentro de esa sala.
El movimiento del personal sigue la dirección del flujo de trabajo y no se permite regresar a salas anteriores dentro de dicho flujo. También considerar el uso de equipo de protección personal (EPP); guantes y batas de laboratorio para diferentes salas los cuales, a menudo, están codificados por colores para facilitar la percepción visual. En cuanto a la gestión de los residuos, éstos deben separarse y eliminarse adecuadamente de acuerdo con las normas de eliminación y control de infecciones aplicables.
La realización de descontaminaciones periódicas es fundamental, cuya frecuencia se puede ajustar si se identifica alguna contaminación, lo que permitirá implementar medidas de descontaminación exhaustivas para resolver rápidamente cualquier problema encontrado. Otra práctica que debe realizarse de manera rutinaria es incluir controles sin plantilla NTC/controles negativos para detectar posible contaminación en los reactivos, consumibles y el entorno general del laboratorio, prestando especial atención a la puesta en marcha y gestión del área de virología molecular como técnicas de amplificación molecular, ya que es una actividad sumamente sensible que fácilmente puede sufrir contaminación.
Calificación de equipos, calibración, mantenimiento y monitoreo
Desde una perspectiva de garantía de calidad, el equipo cubre instrumentos y software, incluida la gestión de información de laboratorio como los sistemas (LIMS); organismos reguladores (ISO 15189 o equivalente) ponen mucho énfasis en el aseguramiento de la calidad de equipos desde la compra inicial hasta la calificación, calibración, mantenimiento y monitoreo, así como su eventual disposición para su uso previsto y ciclo de vida.
Calificación de equipos
El laboratorio de virología clínica contará con un plan de validación y/o verificación de equipos y la fase inicial del mismo será la calificación del equipo, momento en el que el laboratorio evaluará el equipo en consulta con el fabricante con el fin de definir tanto las especificaciones funcionales y operativas del equipo como el coste y eventuales requisitos de instalación, calibración y capacitación. Este paso a menudo se denomina calificación de diseño (DQ) y va seguido de la calificación de instalación (IQ) en la que el laboratorio establece que el equipo es apto para su funcionamiento y cumple con las especificaciones del fabricante. La calificación operativa (OQ) es el tercer paso, el cual implica probar el rendimiento del equipo dentro del entorno clínico, generalmente frente a un “estándar de oro” o método comparador y la fase final es la calificación del desempeño (PQ), donde el desempeño del equipo se monitorea a lo largo del tiempo en el entorno clínico frente a un conjunto definido de criterios de desempeño, normalmente con el apoyo de algunas guías y protocolos que permitan evaluar los parámetros críticos del equipo.
Para los equipos más genéricos utilizados en el laboratorio de virología como pipetas, vórtices y microcentrífugas, las recomendaciones proporcionadas por el fabricante del equipo suelen ser suficientes para calificar dichos equipos en un entorno de rutina.
Particularmente para los ensayos desarrollados en el laboratorio, donde la plataforma de extracción, el kit, la plataforma de amplificación molecular y el kit de detección se combinan en un flujo de trabajo molecular, el laboratorio de virología definirá los criterios de calificación del equipo de acuerdo con las recomendaciones de los fabricantes.
El objetivo es calificar el equipo de laboratorio y monitorear el desempeño para garantizar la validez de datos/resultados generados para los elementos individuales del equipo, así como para todo el flujo de trabajo de prueba.
Calibración
Cuando se utilizan equipos de laboratorio con fines de medición, ya sea para monitorear la carga viral o la temperatura del medio ambiente, mantener el aseguramiento de la calidad del equipo a través de la calibración es esencial. La calibración minimiza la incertidumbre y ayuda a establecer criterios de desempeño operativo que se utilizan para monitorear y controlar errores, garantizando que las mediciones siguen siendo apropiadas y aceptables.
Dentro del laboratorio de virología clínica, la calibración de equipos abarca tanto la planta fija como la maquinaria utilizada para el control de los diferentes entornos de trabajo dentro del laboratorio, así como la instrumentación especializada utilizada en cada una de las áreas, esta actividad generalmente la realiza el fabricante del equipo o un contratista externo con trazabilidad a una norma internacional como ISO 17025 o equivalente.
La frecuencia de calibración depende de los requisitos específicos del equipo, registro de mantenimiento y seguridad, plan de servicio anual, así como el alcance de su uso. Los eventos de calibración se registran dentro de un “registro de equipo/calibración” que identifica el equipo por su número de activo asignado.
Mantenimiento y monitoreo
La calibración, el servicio y el mantenimiento preventivo continuos de los equipos de laboratorio durante toda su vida útil respaldan la confiabilidad y confianza en el rendimiento del equipo. El mantenimiento y el monitoreo deben incluir un registro de todos los equipos indicando números de serie, números de activos asignados y su ubicación dentro del laboratorio.
El resultado de cualquier inspección de seguridad se registra junto con cualquier falla del equipo y eventos de servicio, para lo cual, la mayoría de los laboratorios asignan la responsabilidad de equipos de laboratorio específicos a un operador o grupo de operadores.
Los mantenimientos preventivos, permiten extender lo más posible la vida útil del equipo, posibilitan al laboratorio tomar medidas inmediatas y ayuda con la identificación de tendencias a lo largo del tiempo.
Auditorías internas y externas
Las auditorías internas y externas son una parte esencial del arsenal de garantía de calidad del laboratorio de virología clínica, cuyo propósito es proporcionar una visión general objetiva del control del laboratorio y la eficacia funcional de sus políticas, procesos y procedimientos en relación con el servicio de pruebas que proporciona y el estándar regulatorio, así como el entorno en el que opera.
Las auditorías internas sólo deben ser realizadas por aquellos miembros del personal que estén capacitados y demuestren ser auditores competentes. Además, los miembros del personal que están siendo auditados deben asegurarse de que estén completamente preparados antes de la auditoría con una comprensión completa de las políticas y procedimientos de los que son responsables. La auditoría debe ser no directiva y permitir a los auditados responsables explicar sus procesos y procedimientos. El auditor puede entonces obtener una comprensión de la competencia del personal, identificar áreas en las que se pueden requerir mejoras como capacitación adicional, así como riesgos potenciales o no conformidades dentro de la política y los procedimientos escritos que, en última instancia, podrían conducir a un incumplimiento de la norma regulatoria.
La auditoría externa es una inspección realizada de forma independiente, generalmente por un organismo nacional de acreditación (NAB), que evalúa la competencia técnica del laboratorio y la conformidad con las normas internacionales aplicables, como ISO 15189 o ISO 17025. Las auditorías normalmente siguen un ciclo de cuatro años con una auditoría de seguimiento anual durante un período de tres años y una auditoría de reacreditación cada cuatro años.
Verificación y validación de los ensayos
La implementación de un nuevo método de ensayo o la modificación de un método de ensayo existente debe estar respaldada por un estudio de verificación y/o validación apropiada antes del uso rutinario. La verificación del ensayo es el proceso de prueba y revisión del desempeño de un ensayo en relación con su desempeño conocido o informado, en comparación con las especificaciones de rendimiento definidas por los fabricantes. Considerando que la validación del ensayo es el proceso de evaluar el método de ensayo, establecer sus características de desempeño y determinar su idoneidad para su uso dentro del laboratorio, los requisitos de verificación y validación dependen del tipo de ensayo, su uso previsto y la cantidad de datos de rendimiento disponibles, como los de estudios clínicos anteriores.
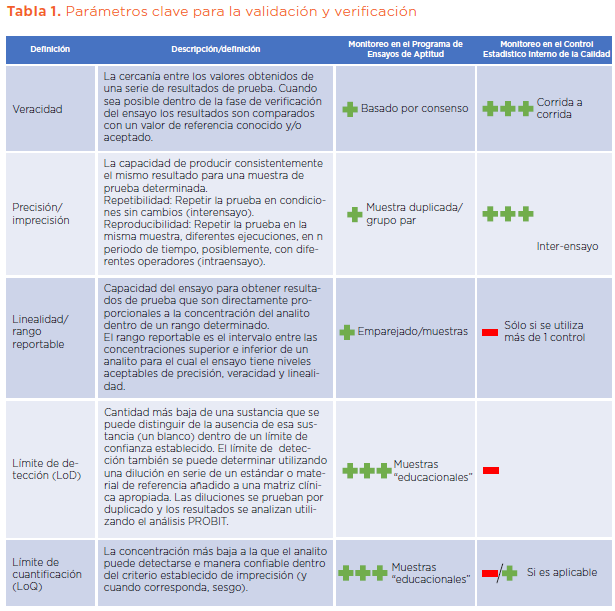
Existen numerosas directrices regulatorias nacionales e internacionales que describen el proceso de verificación y validación, ya que en muchos países esto se rige por los requisitos de las normas regulatorias internas y la categoría regulatoria del método de ensayo, sin embargo, los parámetros generales que deben ser cubiertos son: veracidad, precisión, rango reportable, linealidad (para ensayos cuantitativos), intervalo de referencia, sensibilidad y especificicidad clínicas como se describe en la Tabla 1.
Es importante destacar que, si el laboratorio modifica el ensayo comercial y/o las instrucciones para el uso previsto, el ensayo ya no se considera un ensayo aprobado reglamentariamente y se esperaría que el laboratorio valide completamente el impacto de estos cambios en el rendimiento del ensayo.
El material utilizado para la verificación y validación debe ser de la matriz apropiada y en niveles clínicamente apropiados (si se conocen). En caso de que el material de referencia sea limitado o inexistente, se puede optar por material proveniente del paciente, el cual debe estar adecuadamente caracterizado y, a su vez, se debe considerar utilizar los mismos lotes de reactivos de ensayo al realizar el ejercicio de verificación y validación para reducir el potencial impacto de la variación introducida a través de diferentes lotes. Finalmente, antes de realizar cualquier estudio de verificación o validación, es importante que el laboratorio garantice que el método del ensayo esté calibrado según los requisitos del fabricante y que no haya eventos de recalibración/mantenimientos programados durante el estudio.
Para la verificación de un ensayo cualitativo aprobado reglamentariamente, se aplicarían el enfoque y principios presentados en la Tabla 1.
Veracidad
Para verificar la veracidad se puede realizar un estudio de comparación de métodos. Muestras de pacientes conocidos que cubren el rango del ensayo y se han probado previamente con el método de ensayo actual que utiliza el laboratorio de virología clínica, que a su vez se prueban con el nuevo método propuesto durante un período de tiempo definido y se comparan los resultados. Una alternativa es utilizar los materiales de control o de referencia caracterizados que contienen el objetivo de interés en todo el rango del ensayo.
Los datos cualitativos (positivos, negativos) obtenidos se analizan estadísticamente mediante el análisis K de Cohen.
Precisión
La precisión del método de ensayo se puede determinar probando uno o dos controles en niveles clínicamente relevantes (si se conocen) repetidamente durante un período de tiempo específico para establecer la precisión entre ejecuciones. El tiempo mínimo recomendado es de 20 días en muchas pautas, sin embargo, el intervalo de prueba debe ser suficiente para cubrir la operación de rutina dentro del laboratorio, así como la rotación de operadores.
Los resultados se analizan utilizando la desviación estándar y el coeficiente de variación, y se comparan con las especificacioes del fabricante del ensayo.
Rango reportable y límite de detección (LoD)
El rango reportable del ensayo se verifica utilizando muestras clínicas positivas caracterizadas conocidas en niveles clínicamente relevantes, mediante diluciones de estándares disponibles comercialmente o materiales de referencia probados durante un período de tiempo en comparación con el actual método del laboratorio. El LoD del ensayo se verifica analizando muestras replicadas, por duplicado, en niveles conocidos por encima, en y por debajo del LoD determinado o según lo especificado en el manual de instrucciones del fabricante durante un período definido de tiempo (generalmente 20 días). Luego se calcula el porcentaje detectado en cada nivel y se utiliza el análisis Probit para estimar el LoD.
Para la verificación de un ensayo cuantitativo, los mismos principios básicos utilizados para verificar un ensayo cualitativo se aplican a la veracidad y precisión, aunque es necesario verificarlos en todo el rango de medición analítica (RAM) del ensayo y, en particular, a concentraciones conocidas clínicamente relevantes.
Linealidad (rango analítico de medición)
En el contexto de un ensayo de carga viral, el rango analítico medible (RAM) o rango notificable, se puede definir como el rango lineal de valores de prueba sobre los cuales el laboratorio puede detectar con precisión el ácido nucleico viral objetivo dentro de grados de variación aceptables. Más allá del límite indicado por el fabricante de cuantificación (LoQ), la relación entre la concentración de carga viral medida y real puede no ser lineal y los resultados de la prueba pueden volverse no confiables.
Para este experimento, como mínimo, se deben incluir muestras en los puntos bajo, medio y alto del RAM; el análisis de regresión polinómica se utiliza para evaluar la linealidad del ensayo en todas las concentraciones relevantes y si la relación determina que no son lineales (no de primer orden), esas concentraciones se eliminan del análisis en el extremo superior y/o inferior.
En general, la RAM debe verificarse de acuerdo con los siguientes criterios:
• En cambios de lotes de reactivos, a menos que el laboratorio pueda demostrar que el uso de lotes diferentes no afecta la precisión de resultados de las pruebas del paciente y el rango utilizado para informar resultados.
• Si los materiales de control de calidad reflejan una tendencia o cambio inusual o están fuera de los límites aceptables del laboratorio.
• Después de un mantenimiento o servicio importante del equipo.
• Cuando lo recomiende el fabricante del ensayo.
Sensibilidad y especificidad clínicas
La sensibilidad clínica del método, es la capacidad de detectar la sustancia objetivo en la más mínima concentración o presencia, lo cual puede tener un impacto importante en la precisión del resultado de la prueba y su interpretación en el entorno clínico. Por lo tanto, la clínica del laboratorio de virología debe considerar esto durante la fase de verificación y validación, que es un proceso que continúa mientras el método de ensayo se utiliza en el laboratorio y requiere vigilancia y revisión constantes, así como reevaluación cuando sea requerida. La especificidad, incluye cualquier posible sustancia que interfiera o tenga reacción cruzada y que pueda estar presente en la muestra o matriz. La interferencia es básicamente cualquier organismo o sustancia no objetivo que pueda causar un resultado falso positivo. La lista de posibles sustancias que interfieren y tienen reacciones cruzadas suele estar documentada en el manual de instrucciones del fabricante.
Las pautas del CLSI (EP12-A2) recomiendan un mínimo de 50 muestras clínicas positivas y 50 negativas. En el caso de enfermedades no prevalentes puede resultar difícil obtener la cantidad suficiente para realizar una evaluación clínica completa. En estas circunstancias el laboratorio clínico debe considerar enfoques alternativos, como incluir materiales utilizados en competencias anteriores de desafíos del esquema de pruebas/EQA o materiales comerciales que ya se encuentran caracterizados.
1.- Baylis, S., 2020. Quality assurance and laboratory accreditation In: Encyclopaedia of Virology, fourth ed. Elsevier Science.
2.-Baylis, S.A., Wallace, P., McCulloch, E., et al., 2019. Standardization of nucleic acid tests: The approach of the World Health Organization. Journal of Clinical Microbiology 57.
3.-Bustin, S.A., Benes, V., Garson, J.A., et al., 2009. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry 55 (4).
4.-EP05-A3E. Evaluation of Precision of Quantitative Measurement Procedures, third ed. Clinical Laboratory Standard Institute (CLSI).
5.-Huggett, J.F., Foy, C.A., Benes, V., et al., 2020. The digital MIQE guidelines update: Minimum information for publication of quantitative digital PCR experiments for 2020-Clinical Chemistry 66 (8), 1012–1029.
6.- Hayden, R., Sun, Y., Tang, L., et al., 2017. Progress in quantitative viral load testing: Variability and impact of the WHO Quantitative International Standards. Journal of Clinical Microbiology 55 (2).
7.- ISO17511, 2020. In vitro diagnostic medical devices – Requirements for establishing metrological traceability of values assigned to calibrators, trueness control materials and human samples.
8.- ISO17043, 2010. Conformity assessment – General requirements for proficiency testing.
9.- Miller, M.B., Atrzadeh, F., Burnham, C.D., et al., 2019. Clinical utility of advanced microbiology testing tools. Journal of Clinical Microbiology 57 (9). doi:10.1128/JCM.00495-19.
10.- Whale, A.S., Jones, G.M., Pavšicˇ, J., et al., 2018. Assessment of digital PCR as a primary reference measurement procedure to support advances in precision medicine. Clinical Chemistry 64 (9), 1296–1307.