Generalidades
La hemofilia A y B son trastornos hemorrágicos hereditarios con un patrón recesivo ligado al cromosoma X, caracterizados por la deficiencia parcial o completa de los factores de coagulación circulantes VIII (FVIII) o IX (FIX), respectivamente. Los varones se ven afectados predominantemente por tener un solo cromosoma X. La característica distintiva de la hemofilia grave es la presencia de episodios hemorrágicos recurrentes, espontáneos, prolongados y anormales que afectan principalmente a los tejidos blandos y las articulaciones sinoviales1.
Diagnóstico
El médico realiza una historia familiar completa que explora las posibles manifestaciones de sangrado en otros miembros de la familia, sin olvidar que puede ocurrir una mutación de novo esporádica. Cuando se sospecha hemofilia, la evaluación de laboratorio inicial debe incluir un hemograma completo, tiempo de protrombina, TTPa, estudios de mezcla en caso de TTPa prolongado, un nivel de fibrinógeno y un antígeno vWF y nivel de actividad. Los niños con hemofilia presentan un TTPa prolongado aislado y un recuento de plaquetas y tiempo de protrombina normales. Los pacientes con hemofilia grave suelen tener un TTPa 2 o 3 veces mayor al límite superior de la normalidad. A menos que el paciente tenga un inhibidor activo de FVIII o FIX, el estudio de mezcla de TTPa se corregirá con la adición de plasma normal. Se deben cuantificar los niveles de actividad de FVIII o FIX de manera específica para confirmar el diagnóstico y diferenciar de otros trastornos hemorrágicos hereditarios. Las pruebas genéticas son una parte importante de la evaluación diagnóstica de la hemofilia; ya que permiten un asesoramiento genético preciso y reconocen mutaciones asociadas con el riesgo potencial de desarrollo de inhibidores1-3.
Tratamiento
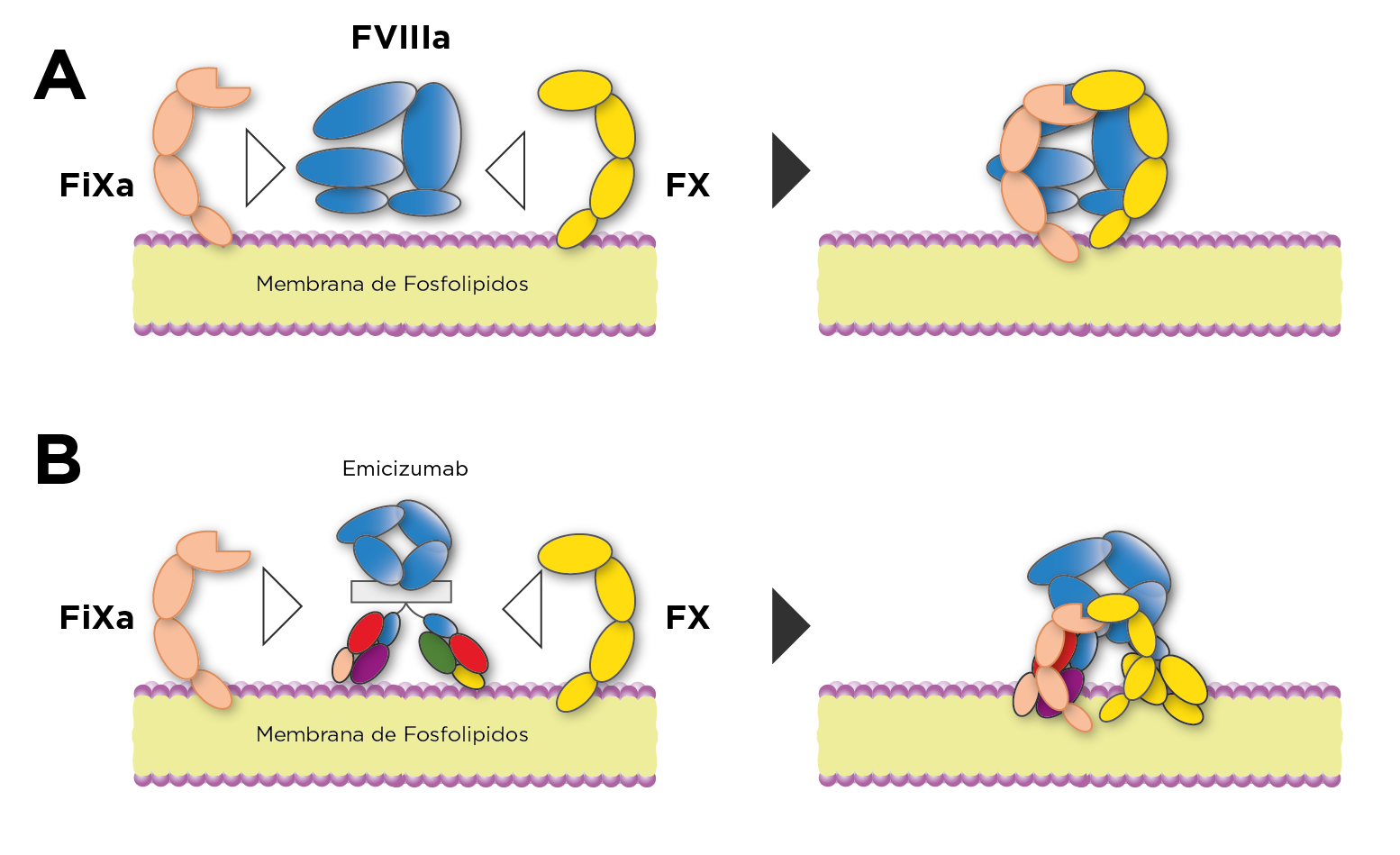
Desde que se entendió que la falta de un factor de la coagulación era la causa del sangrado en hemofilia, se planteó la necesidad de sustituirlo, iniciando con el uso de sangre y plasma humano, seguido a través del tiempo con: crioprecipitados, factores purificados del plasma, factores de origen recombinante, factores con vida media extendida, hasta llegar en esta última década a la terapia sin factor sustitutivo y la terapia génica. Son varias las moléculas de FVIII y FIX que quedan en el rubro de factores de vida media extendida producidas por diferentes farmacéuticas. Dentro de las terapias sin factor sustitutivo, la más utilizada es emicizumab, un anticuerpo biespecífico monoclonal humanizado desarrollado para unir FIX y FX activados en la membrana de fosfolípidos imitando la funcionalidad del cofactor FVIII (figura 1).
Otro fármaco utilizado es Fitusiran, un ARN de interferencia pequeño en investigación diseñado para la supresión de la antitrombina a través del silenciamiento génico postranscripcional en los hepatocitos, lo que aumenta la cantidad de generación de trombina. El concizumab es un anticuerpo monoclonal humanizado en fase de investigación que bloquea la acción reguladora fisiológica del TFPI (inhibidor de la vía del factor tisular), incrementando la generación de trombina en pacientes con hemofilia e individuos sanos. La cura definitiva ha sido durante mucho tiempo el sueño de los pacientes con hemofilia, con la terapia génica está cada vez más cercana1-5.
Monitorización del tratamiento
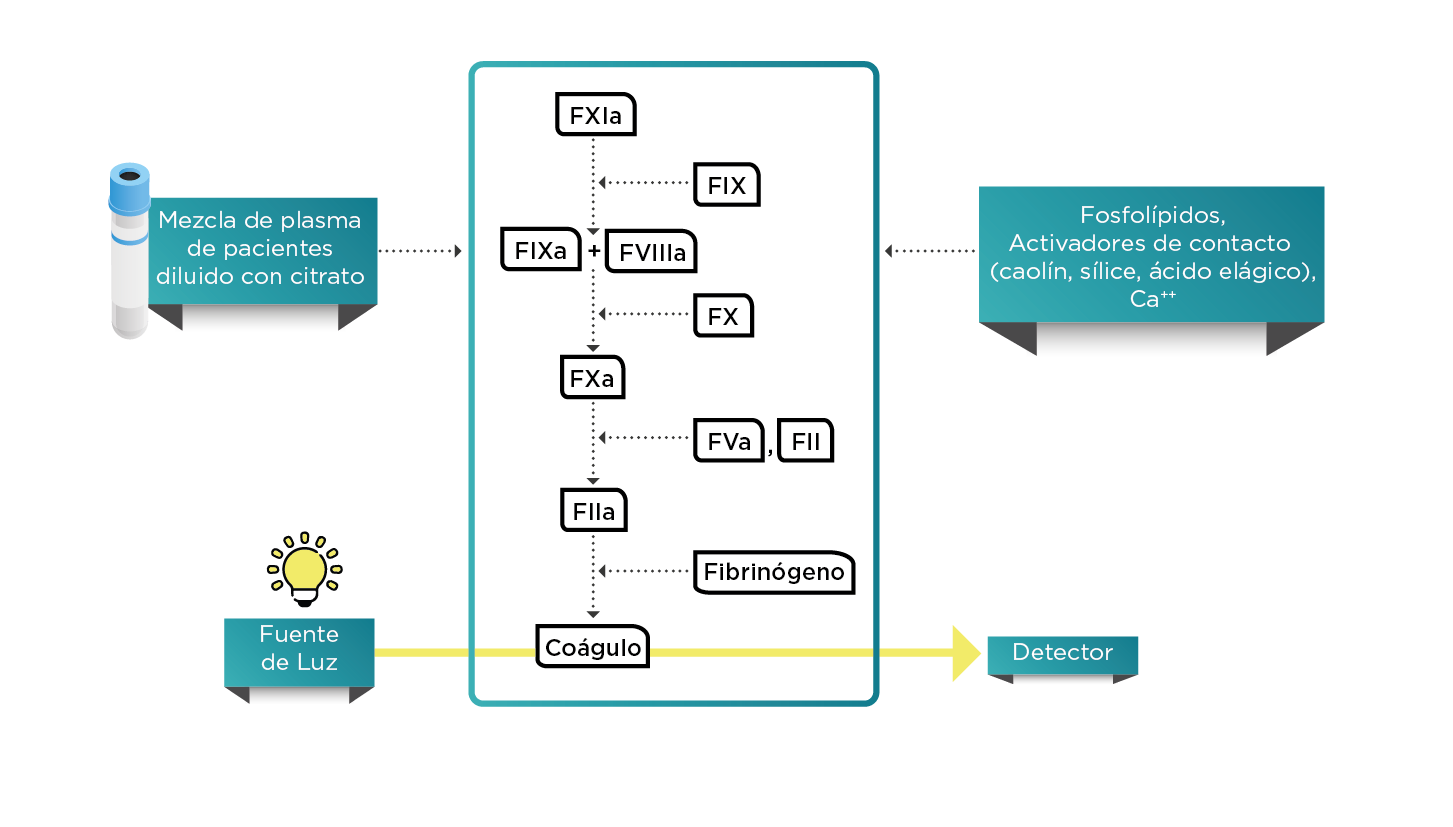
A los pacientes con hemofilia se les realiza un seguimiento mediante pruebas de laboratorio para evaluar el grado de recuperación del factor después de la infusión de concentrados por profilaxis o durante una cirugía de riesgo. Tradicionalmente, el tratamiento con concentrados de factor de coagulación ha sido (y aún es) evaluado a través de la medición del factor de coagulación de interés después de la infusión por medio del ensayo de coagulación de una etapa, que se basa en el TTPa y el plasma deficiente en factor, en la figura 2 se muestra un diagrama del ensayo de coagulación de una etapa para FVIII y FIX. El principio y reactivos son los mismos, pero difieren en el factor deficiente (FVIII o FIX).
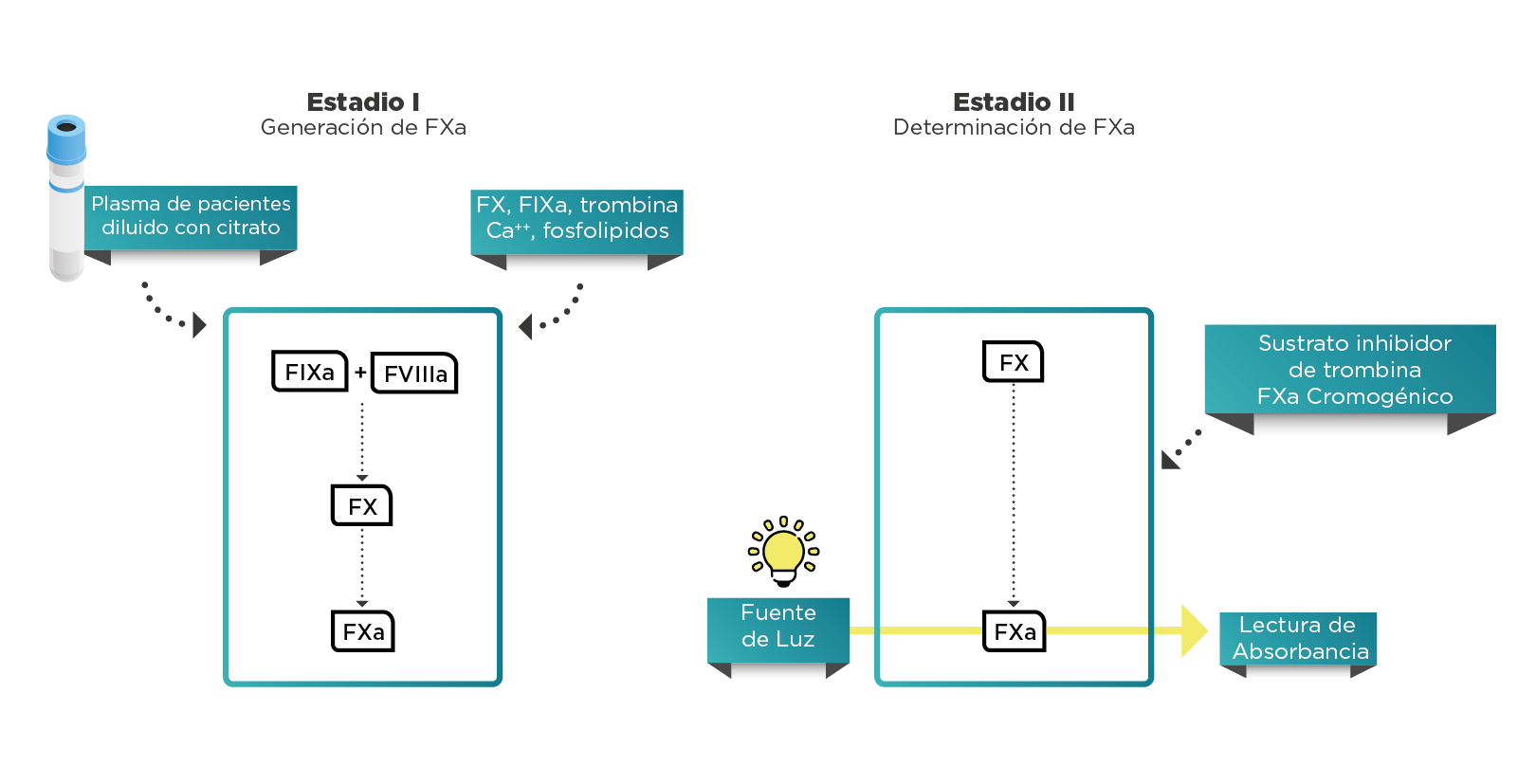
Estos factores también se pueden medir por medio de otra clase de ensayos basados en la tecnología de sustratos cromogénicos sintéticos (figura 3).
La diferencia más importante entre los ensayos cromogénicos y de coagulación de una etapa radica en el hecho de que, en el último, la coagulación se activa a partir de los factores de contacto hasta el FXa. El FXa generado, que (bajo las condiciones del ensayo) depende de FVIII o FIX, a su vez se mide mediante un sustrato cromogénico sintético específico para FXa (Figura 3) y no mediante la detección de coágulos como ocurre con el ensayo de coagulación de una etapa1-5.
Actualmente, no existe un consenso sobre el grado de diferencia en los resultados para un mismo producto entre los distintos ensayos, ni tampoco del resultado con lo que marca la ficha técnica del fabricante. No utilizar métodos que se alejen más de un 25-30% en los resultados obtenidos respecto a lo reportado en las fichas técnicas. Los factores de derivados plasmáticos para hemofilia A o B se recomienda el uso del FVIII:C o FIX:C en una etapa o del FVIII o FIX cromogénico de forma indistinta. Para los factores recombinantes completos no modificados, en el caso de la hemofilia A se recomienda el FVIII:C de una etapa o el FVIII cromogénico para su monitorización. En el caso de la hemofilia B, en pacientes tratados con FIX recombinante no modificado la técnica de elección será el FIX por método coagulométrico. Para los factores recombinantes modificados en Hemofilia A existen recomendaciones publicadas por la Federación Mundial de Hemofilia pues las moléculas son muy variadas, de la mayoría de ellos puede monitorizarse su eficacia con los métodos cromogénicos y en menor proporción, con los métodos basados en el TTPa o coagulométricos1-6.
Respecto a la monitorización de tratamientos no sustitutivos en hemofilia, el fármaco más utilizado es el emicizumab, un anticuerpo biespecífico diseñado que se une tanto al FIX/FIXa como al FX/FXa y que no se rige por los mecanismos que regulan el FVIII, pero actúa como un FVIII mimético. El TTPa y todas las determinaciones derivadas se ven acortados o sobrestimados por este anticuerpo, por lo que no son útiles para su monitorización. Emicizumab interfiere en las determinaciones cromogénicas del FVIII que utilizan FIXa y FX humanos, pero no en los que utilizan FIXa y FX de origen bovino por lo que para medir la actividad de FVIII endógeno en pacientes en tratamiento con emicizumab se recomienda usar FVIII cromogénico que contenga FX de origen bovino. Consecuentemente, para tipificar y cuantificar los inhibidores en pacientes con hemofilia A tratada con emicizumab, se debe utilizar en el ensayo Bethesda reactivo de FVIII cromogénico de origen bovino. En la actualidad, para los pacientes sometidos a terapia génica, no existe evidencia científica sólida ni de consenso sobre los ensayos de laboratorio más adecuados para monitorizar este tipo de terapia en hemofilia A o B1-6.
Conclusiones:
Es importante tener un conocimiento bioquímico de las propiedades de las nuevas terapias para la interpretación de los resultados obtenidos. También es importante comprender cómo las nuevas terapias afectan otros ensayos clínicos. El tratamiento de la hemofilia está entrando en una nueva era y su monitorización por laboratorio debe seguir en paralelo.
Bibliografìa1. Matuk-Villazon O, Roberts JC, Corrales-Medina FF. Hemophilia: The Past, the Present, and the Future, Pediatrics in Review 2021; 42(12): 672–683
2. Tripodi A, Chantarangkul V, Novembrino C, Peyvandi F. Clin Chem 2019; 65(2): 254 – 262.
3. Kitchen S, Tiefenbacher S, Gosselin R. Factor activity assays for monitoring extended half-life FVIII and factor IX replacement therapies. Semin Thromb Hemost 2017; 43(3): 331–337.
4. Al-Samkari H, Croteau SE. Shifting landscape of hemophilia therapy: Implications for current clinical laboratory coagulation assays. Am J Hematol 2018; 93: 1082–1090.
5. Lenting PJ. Laboratory monitoring of hemophilia A treatments: new challenges. Blood Advances 2020; 4(9): 2111 – 2118.
6. https://guidelines.wfh.org/chapter/laboratory-diagnosis-and-monitoring/